Key Points
PMos scavenge endothelial adherent sickle RBCs in vitro and in vivo.
Vaso-occlusion in sickle mice can be controlled through manipulation of PMo numbers.

Abstract
Painful vaso-occlusive crisis (VOC) is the most common complication of sickle cell disease (SCD). Increasing evidence suggests that vaso-occlusion is initiated by increased adherence of sickle red blood cells (RBCs) to the vascular endothelium. Thus, the mechanisms that remove endothelial-attached sickle RBCs from the microvasculature are expected to be critical for optimal blood flow and prevention of VOC in SCD. We hypothesized that patrolling monocytes (PMos), which protect against vascular damage by scavenging cellular debris, could remove endothelial-adherent sickle RBCs and ameliorate VOC in SCD. We detected RBC (GPA+)-engulfed material in circulating PMos of patients with SCD, and their frequency was further increased during acute crisis. RBC uptake by PMos was specific to endothelial-attached sickle, but not control, RBCs and occurred mostly through ICAM-1, CD11a, and CD18. Heme oxygenase 1 induction, by counteracting the cytotoxic effects of engulfed RBC breakdown products, increased PMo viability. In addition, transfusions, by lowering sickle RBC uptake, improved PMo survival. Selective depletion of PMos in Townes sickle mice exacerbated vascular stasis and tissue damage, whereas treatment with muramyl dipeptide (NOD2 ligand), which increases PMo mass, reduced stasis and SCD associated organ damage. Altogether, these data demonstrate a novel mechanism for removal of endothelial attached sickle RBCs mediated by PMos that can protect against VOC pathogenesis, further supporting PMos as a promising therapeutic target in SCD VOC.
Introduction
Painful vaso-occlusive crisis (VOC) is the most common complication of sickle cell disease (SCD) with repeated episodes of VOC contributing to morbidity and multiorgan injury.1-3 A major contributor to microvascular occlusion in SCD is adherence of sickle red blood cells (RBCs) to the underlying vascular endothelial cells (ECs).4-6 Indeed, the extent of sickle RBC adhesiveness is an important biomarker that correlates with the severity of clinical VOC.7 Once deoxygenated, EC-attached sickle RBCs are resistant to removal from the vascular merely by flow shear forces.8 Attachment of sickle RBCs activates the underlying endothelium and, together with other blood components, leads to vaso-occlusion.9-12 Intravascular hemolysis, also a hallmark of SCD, further activates the vascular endothelium, increasing the attachment of sickle RBCs to the vessel wall.13-16 Haptoglobin and hemopexin, which normally remove free hemoglobin and heme, respectively, from the circulation, are depleted in patients with SCD.17-19 Heme oxygenase 1 (HO-1) is a critical heme detoxification enzyme that degrades heme and also confers cytoprotective, anti-inflammatory and antioxidant effects through the heme breakdown products carbon monoxide and biliverdin.20,21 We recently showed that nonclassical circulating patrolling monocytes (PMos), which play a key role as housekeepers of the vasculature, express high levels of HO-1 (HO-1hi) in patients with SCD and that HO-1hi PMo numbers were lower in patients with recent or recurrent pain episodes.22 We also found that PMos scavenge heme-damaged ECs and thus protect against hemolysis-induced endothelial damage.22,23 These data have led to the proposal that along with hemopexin and haptoglobin, PMos are an important line of defense against hemolytic damage to the endothelium.24
RBC transfusions remain a cornerstone treatment in the management of SCD complications with close to 90% of patients needing at least one transfusion in their lifetime.25,26 Transfusions correct anemia, suppress ineffective erythropoiesis, lower the percentage of circulating sickle RBCs, and decrease blood viscosity.27-29 Whereas some indications for transfusion are strongly recommended and supported by randomized controlled data, such as for stroke prevention, others remain controversial, such as transfusion for prevention of recurrent episodes of VOC.30 Limited mechanistic understanding of diverse disease complications in SCD and paucity of data on detailed cellular mechanisms of improvement with transfusion are additional factors that have hampered determination of appropriate transfusion indications in SCD.
In this study, we tested the hypothesis that PMos, which are highly effective in clearance of endothelium-attached materials,31-33 might scavenge endothelial-attached sickle RBCs and protect against VOC. We further reasoned that in steady state (non-VOC), RBC transfusions by diluting sickle RBC mass would decrease RBC uptake by PMos, thus prolonging their survival to effectively clear the vasculature in SCD.
Methods
Detailed experimental procedures are outlined in supplemental Data, available on the Blood Web site. Briefly, characterization of glycophorin A (GPA)–expressing engulfed cells was performed on blood samples obtained from patients with SCD after informed consent. For in vitro studies, human lung microvascular endothelial cells (HMVECs) were pretreated with hemin and cocultured with carboxyfluorescin diacetate succinimidyl ester (CFSE)–labeled RBCs and purified monocytes before analysis by flow cytometry (FCM) and ImageStream. For in vivo studies, 1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate (Dil)-labeled RBCs (1.5 × 109 cells) were injected first, followed by hemin (30 μmol/kg), into Nr4a1-GFP+/− mice (Tg(Nr4a1-EGFP/cre)820Khog). Quantification of DiI-labeled RBC uptake by GFP+ PMos in the vasculature was performed following perfusion by whole-mount imaging using confocal microscopy. Townes-SS mice (Hbb hAγβS/hAγβS) were treated with muramyl dipeptide (MDP) to induce PMos34 or with a low dose of liposome-encapsulated dichloromethylene-bisphosphonate (Clo-Lipo) to selectively deplete PMos.35,36 Stasis was then assessed by quantifying i) Ter-119 positive RBCs in the vasculature using immunofluorescence and ii) the percentage of occluded vessels by histology using hematoxylin and eosin (H&E) staining. Data are represented as mean ± standard error of the mean (SEM). Statistical significance of the differences between experimental groups was determined by two-tailed Student t test; P < .05 was considered statistically significant.
Data sharing statement
Data are provided in the supplemental Materials and methods.
Results
Circulating PMos from patients with SCD express RBC-derived material
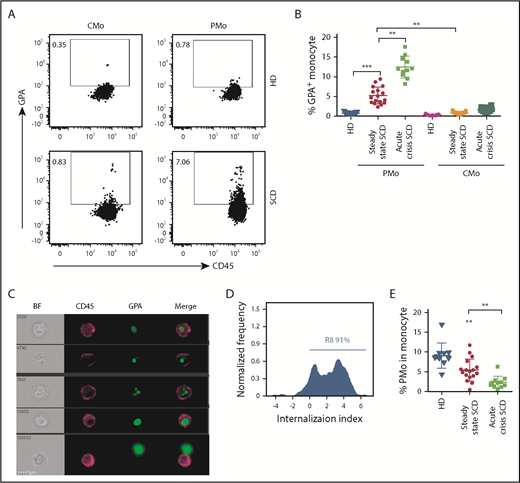
To test the hypothesis that circulating PMos from patients with SCD phagocytose sickle RBCs, we checked for the presence of RBC-derived material intracellularly in monocytes by FCM. Patients’ relevant clinical information is shown in supplemental Table 1. As compared with race-matched healthy donors (HDs), PMos from patients with SCD at steady state (non-VOC) expressed higher GPA levels (5% ± 0.5% vs 0.7% ± 0.04%, P < .001), or SCD classical monocytes (CMos: 0.8% ± 0.07%, P < .01; Figure 1A-B). Band 3 (another RBC marker) had a similar expression pattern in SCD and HD monocytes as GPA (supplemental Figure 1A-B). Patients with SCD experiencing acute VOC presented with even higher GPA+ PMos (13% ± 1%, P < .01) (Figure 1A-B; supplemental Table 2). To discriminate between adherent and internalized RBC materials, we analyzed the samples using imaging flow cytometry (IFC). We found that most GPA+ PMos from patients with SCD (∼91%) had intracellularly localized GPA (Figure 1C-D), indicating that GPA+ RBC materials are indeed internalized by PMos. Overall, the frequency and absolute number of PMos in acute crisis were lower than that in steady-state SCD (Figure 1E; frequencies: 2.4% ± 0.4% vs 5.5% ± 0.7%, P < .01) (supplemental Figure 1C; numbers: 15 ± 3 cells/μL vs 56 ± 8 cells/μL, P < .01), whereas the absolute numbers of CMo were comparable (supplemental Figure 1D). Taken together, these results demonstrate that engulfed RBC material is present in circulating PMos from patients with SCD, but not HDs, and that during acute crisis, RBC-ingested PMo frequency is increased, even though overall PMo numbers decrease.
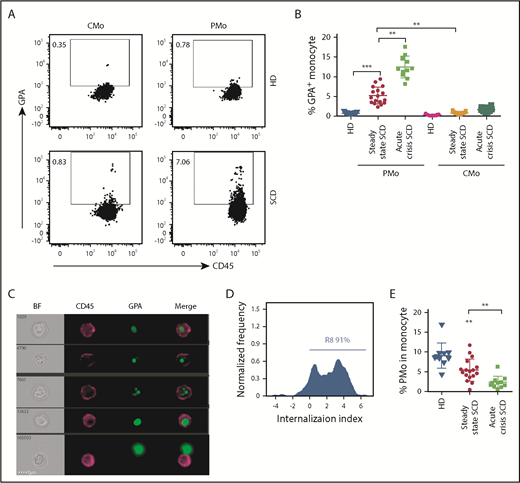
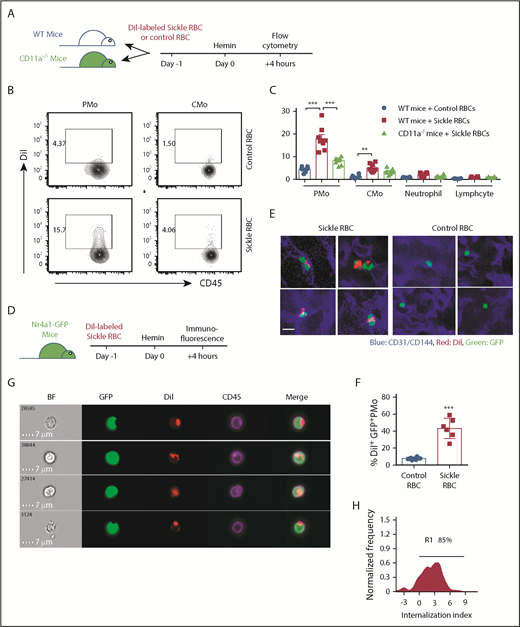
Circulating PMos engulf sickle RBCs in SCD. (A) Representative dot plots comparing GPA+ (an RBC marker) CMos (CD45+HLA-DR+SSChiCD14hiCD16−) and PMos (CD45+HLA-DR+SSChiCD14lowCD16+) from race-matched HDs and patients with SCD. For the gating strategy used, see supplemental Figure 4A. (B) Frequencies of GPA+ circulating monocyte populations in HDs (n = 17), patients with SCD at steady state (n = 17), and during acute crisis (n = 11). (C) Representative IFC images acquired simultaneously and gated on GPA+ PMos from patients with SCD (from A). Left to right: single-channel bright field (BF), CD45 (representing PMos in purple), GPA (representing RBC materials in green), and merged images showing GPA+ materials within CD45+ cells in the top 4 rows, but not in the bottom row, where GPA+ material is attached to the external surface of PMos. (D) Histogram depicting degree of GPA+ material internalized within PMos with internalization score (<0 represents GPA+ material attached to the surface of PMos; >0 represents GPA+ material internalized by PMos). Percentage of cells with an internalization score >0 is indicated. (E) Frequency of PMos within total circulating monocytes from HD (n = 10), patients with SCD at steady state (n = 17), and during acute crisis (n = 11) were determined by FCM. Data represent mean ± SEM; means were compared using a 2-tailed Student t test. **P < .01; ***P < .001.
Circulating PMos engulf sickle RBCs in SCD. (A) Representative dot plots comparing GPA+ (an RBC marker) CMos (CD45+HLA-DR+SSChiCD14hiCD16−) and PMos (CD45+HLA-DR+SSChiCD14lowCD16+) from race-matched HDs and patients with SCD. For the gating strategy used, see supplemental Figure 4A. (B) Frequencies of GPA+ circulating monocyte populations in HDs (n = 17), patients with SCD at steady state (n = 17), and during acute crisis (n = 11). (C) Representative IFC images acquired simultaneously and gated on GPA+ PMos from patients with SCD (from A). Left to right: single-channel bright field (BF), CD45 (representing PMos in purple), GPA (representing RBC materials in green), and merged images showing GPA+ materials within CD45+ cells in the top 4 rows, but not in the bottom row, where GPA+ material is attached to the external surface of PMos. (D) Histogram depicting degree of GPA+ material internalized within PMos with internalization score (<0 represents GPA+ material attached to the surface of PMos; >0 represents GPA+ material internalized by PMos). Percentage of cells with an internalization score >0 is indicated. (E) Frequency of PMos within total circulating monocytes from HD (n = 10), patients with SCD at steady state (n = 17), and during acute crisis (n = 11) were determined by FCM. Data represent mean ± SEM; means were compared using a 2-tailed Student t test. **P < .01; ***P < .001.
PMos uptake sickle RBCs cocultured with ECs
To examine whether PMos can uptake sickle RBCs in vitro, we cocultured purified monocytes from HDs with CFSE-labeled sickle (HbSS) RBCs or HD (HbAA) RBCs and quantified the frequency of CFSE+ material associated with PMos vs CMo using FCM. Unexpectedly, we found comparable low frequencies of CFSE+ monocyte subsets cocultured with HbAA or HbSS RBCs (Figure 2A-C). Since PMo scavenge damaged vascular endothelium,22,32 we reasoned that interaction with ECs was essential for PMo sickle RBC uptake. Indeed, the presence of HMVECs induced a fourfold increase in the frequency of CFSE+ PMos from HD when cultured with sickle RBCs (Figure 2C; 2.4% ± 0.3% to 11.1% ± 0.5%, P < .001). This increase was only seen with sickle RBCs, as there was minimal increase of CFSE+ PMo in cocultures with HMVEC-HD RBCs (Figure 2C; 2.1% ± 0.3% to 2.7% ± 0.4%, P = .39). Pretreatment of HMVECs with hemin further increased the frequency of CFSE+ PMos (Figure 2C; 17.8% ± 0.9%, P < .01). Similarly, pretreatment of HMVECs with tumor necrosis factor α (TNF-α) also increased the uptake of sickle RBCs by PMos (15.8 ± 0.9 vs 10.6 ± 1.4, P < .05, n = 4). We found no or at least minimal effect in the frequencies of CFSE+ CMos from HD under any conditions tested (Figure 2C; P > .05 in all comparisons). We next examined circulating PMos from patients with SCD in their ability to uptake sickle RBC materials (Figure 1) and found that cocultures with HMVECs also resulted in increased SCD CFSE+ PMos (Figure 2D; 2.6% ± 0.3% to 8.7% ± 0.9%, P < .001). In addition, pretreatment of HMVECs with hemin further increased the frequency of CFSE+ PMos from SCD (Figure 2D; 12.9% ± 1.4%, P < .001). Altogether, these data demonstrate that sickle RBCs can be phagocytosed by PMos when in contact with ECs in vitro and that CMos are incapable of RBC uptake in our in vitro culture system.
![Figure 2. PMo uptake sickle RBCs when cocultured with HMVEC. (A) Schematic representation of experimental design. HMVECs were pretreated with hemin or PBS for 2 hours and subjected to 2 washes before the addition of purified total monocytes and CFSE-labeled RBCs. Some monocytes and labeled RBCs were cultured in the absence of HMVECs. After overnight cultures, the uptake of CFSE+ RBCs by monocyte subsets was analyzed by FCM and IFC. (B) Representative dot plots comparing CFSE+ PMos and CFSE+ CMos from cultures shown in panel A. Frequencies of CFSE+ monocyte subsets from HDs (n = 3-12; C) and patients with SCD (n = 6-12; D). (E) HMVECs were preincubated with individual blocking antibodies (anti-ICAM-1, anti-VCAM-1, anti-CD11a, anti-CD11b, anti-P-selectin, anti-PSGL-1, anti-CD11c, anti-CD18, anti-CD31, anti-CD16, anti-CD32, anti-CD64, anti-Fc α/µ receptor, or isotype matched control [mouse IgG] antibodies) or annexin V or tin protoporphyrin IX (SnPPIX) for 30 minutes before the addition of HD monocytes and CFSE-labeled sickle RBCs. It should be noted that it takes ∼10 to 15 minutes for monocytes and RBCs to settle on the HMVEC. Frequencies of CFSE+ PMos were then analyzed (n = 4-12). Data represent mean ± SEM; means were compared using a 2-tailed Student t test. *P < .05; ***P < .001. Ab, antibody.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/7/10.1182_blood.2019000172/3/m_bloodbld2019000172f2.png?Expires=1767708701&Signature=qCFFtIM4xMx2DcklM~JWDWt0mybM19VKi58pkCmnj8YkDAsB6pbGssWoJ9GJQAKqp16cE-FgDsdE0YJxQbuCXPjLSEabV19feacylFT~H5geCk8QCTbBqkCALLdg1rJFMRhzaRAFoIRERxEYpdkesy78xqf4kkCxUDCwTUeAzKNI89uDdTitv0LxEK5Ri1c5GfsM4Ew-X7toYOP-sO4RHbKJDPUR53tT9duC5cG7gEOw63T-0al6DIivi~1u6TRsObHgsBf-~eCUghSXOa2OSSjQ-~7fSvOKqjOAE9cKr0O0tYnmHnYBBtvddV3e7KoeZmnsiBb0mcWsNo6k0fIFiw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PMo uptake sickle RBCs when cocultured with HMVEC. (A) Schematic representation of experimental design. HMVECs were pretreated with hemin or PBS for 2 hours and subjected to 2 washes before the addition of purified total monocytes and CFSE-labeled RBCs. Some monocytes and labeled RBCs were cultured in the absence of HMVECs. After overnight cultures, the uptake of CFSE+ RBCs by monocyte subsets was analyzed by FCM and IFC. (B) Representative dot plots comparing CFSE+ PMos and CFSE+ CMos from cultures shown in panel A. Frequencies of CFSE+ monocyte subsets from HDs (n = 3-12; C) and patients with SCD (n = 6-12; D). (E) HMVECs were preincubated with individual blocking antibodies (anti-ICAM-1, anti-VCAM-1, anti-CD11a, anti-CD11b, anti-P-selectin, anti-PSGL-1, anti-CD11c, anti-CD18, anti-CD31, anti-CD16, anti-CD32, anti-CD64, anti-Fc α/µ receptor, or isotype matched control [mouse IgG] antibodies) or annexin V or tin protoporphyrin IX (SnPPIX) for 30 minutes before the addition of HD monocytes and CFSE-labeled sickle RBCs. It should be noted that it takes ∼10 to 15 minutes for monocytes and RBCs to settle on the HMVEC. Frequencies of CFSE+ PMos were then analyzed (n = 4-12). Data represent mean ± SEM; means were compared using a 2-tailed Student t test. *P < .05; ***P < .001. Ab, antibody.
PMo uptake sickle RBCs when cocultured with HMVEC. (A) Schematic representation of experimental design. HMVECs were pretreated with hemin or PBS for 2 hours and subjected to 2 washes before the addition of purified total monocytes and CFSE-labeled RBCs. Some monocytes and labeled RBCs were cultured in the absence of HMVECs. After overnight cultures, the uptake of CFSE+ RBCs by monocyte subsets was analyzed by FCM and IFC. (B) Representative dot plots comparing CFSE+ PMos and CFSE+ CMos from cultures shown in panel A. Frequencies of CFSE+ monocyte subsets from HDs (n = 3-12; C) and patients with SCD (n = 6-12; D). (E) HMVECs were preincubated with individual blocking antibodies (anti-ICAM-1, anti-VCAM-1, anti-CD11a, anti-CD11b, anti-P-selectin, anti-PSGL-1, anti-CD11c, anti-CD18, anti-CD31, anti-CD16, anti-CD32, anti-CD64, anti-Fc α/µ receptor, or isotype matched control [mouse IgG] antibodies) or annexin V or tin protoporphyrin IX (SnPPIX) for 30 minutes before the addition of HD monocytes and CFSE-labeled sickle RBCs. It should be noted that it takes ∼10 to 15 minutes for monocytes and RBCs to settle on the HMVEC. Frequencies of CFSE+ PMos were then analyzed (n = 4-12). Data represent mean ± SEM; means were compared using a 2-tailed Student t test. *P < .05; ***P < .001. Ab, antibody.
To characterize the molecular requirements for sickle RBC uptake, we performed blocking experiments using antibodies against previously identified molecules important in sickle RBC-EC and/or monocyte interactions.32,37-39 A dose-dependent inhibition of sickle RBC uptake (CFSE+ PMos) up to ∼50% was found with antibodies against ICAM-1, CD11a, or CD18 (P < .001), and a roughly 20% decrease was found with antibodies against VCAM-1, P-selectin, or CD16 (P < .05; supplemental Figure 1E; Figure 2E). No inhibition was detected when antibodies against CD11b, CD11c, CD31, PSGL-1, CD32, CD64, or Fc α/μ receptor were used or when we used a phosphatidylserine inhibitor (annexin V) or tin protoporphyrin IX (SnPPIX, an HO-1 inhibitor) (Figure 2E). Both sickle and HD RBCs expressed extremely low levels of surface immunoglobulin G (IgG; 1.9% ± 0.3% vs 1.7% ± 0.2%), and use of IgG-free media did not affect sickle RBC uptake by PMos (12.6% ± 1.3% vs 12.3% ± 1.6%; supplemental Figure 1F-H), suggesting an IgG-independent mechanism for uptake. Altogether, these data indicate that in the presence of ECs, sickle RBCs can be engulfed by PMos in part through ICAM-1, CD11a, and CD18.
PMos uptake sickle RBCs in vivo
We next examined PMo uptake of sickle RBCs in vivo. DiI-labeled RBCs from Townes SCD mice or litter-matched non-SCD (AA) control mice were transfused into recipient wild-type (WT) mice followed by injection with hemin. Consistent with the patient data (Figures 1 and 2), a higher frequency of DiI+ PMos (CD11b+CD115+Ly-6C−) was present in mice that had been transfused with sickle RBCs (13.5% ± 1.2%) as compared with mice transfused with control RBCs (Figure 3A-C; 4.7% ± 0.2%, P < .01). All other circulating blood leukocytes exhibited background levels of DiI+ cells in both groups of mice, although we detected a marginal increase in DiI+ CMos of mice that had received sickle RBCs (Figure 3C; 3.7% ± 0.4%, P < .01). We also transfused sickle RBCs into CD11a−/− mice and found a decrease (by 50% [P < .05]) in the frequency of DiI+ PMos as compared with that of sickle RBC–transfused WT mice (Figure 3C), confirming a role for CD11a in PMo uptake of sickle RBCs.
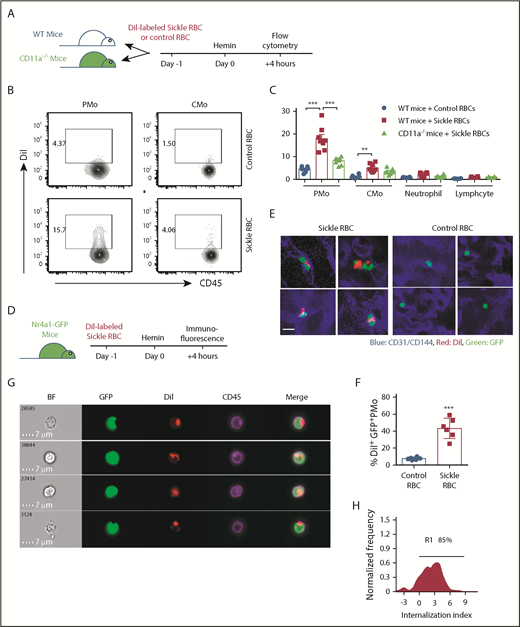
PMos uptake sickle RBCs in vivo. (A) Schematic representation of experimental design. WT mice or CD11a−/− mice were transfused with DiI-labeled RBCs from sickle Townes or control HbAA mice. At 20 hours after transfusion, mice were injected IV with freshly prepared hemin and analyzed 4 hours later. (B) Representative dot plots comparing DiI+ PMos (CD45+CD11b+Ly-6G-CD115+Ly-6C−) and CMo (CD45+CD11b+Ly-6G-CD115+Ly-6C+) from WT mice. For the gating strategy used, see supplemental Figure 4B. (C) Frequencies of DiI+ circulating leukocyte subsets in WT mice (n = 8) and CD11a−/− mice (n = 8). (D) Schematic representation of experimental design; Nr4a1-GFP mice received treatment as shown in panel A and were analyzed with immunofluorescence following perfusion with PBS to wash out nonattached circulating blood cells. (E) Representative immunofluorescence images showing the uptake of DiI+ RBCs by GFP+ PMos on the endothelial surface of liver and lung in mice that received DiI-labeled sickle RBCs or control RBCs. CD31/CD144 (endothelial markers, blue), DiI (RBC in red), and GFP (PMo, green). Scale bar, 5 µm. (F) The frequencies of GFP+ PMos with uptake of DiI+ RBC (DiI+ GFP+ PMos) in mice as shown in panel E. (G) Representative IFC images acquired and gated on DiI+ PMos from hemin-treated Nr4a1-GFP reporter mice. Left to right: single-channel BF, GFP (representing PMos in green), DiI (representing sickle RBC material in red), CD45 (representing surface PMos in purple), and merged images showing DiI+ materials within GFP+CD45+ PMos. (H) Histogram depicting degree of DiI+ material internalized within GFP+ PMos with internalization score (<0 represents DiI+ material attached to the surface of PMo; >0 represents DiI+ material internalized by PMos). Percentage of cells with an internalization score >0 is indicated. Data represent mean ± SEM; means were compared using a 2-tailed Student t test. **P < .01; ***P < .001.
PMos uptake sickle RBCs in vivo. (A) Schematic representation of experimental design. WT mice or CD11a−/− mice were transfused with DiI-labeled RBCs from sickle Townes or control HbAA mice. At 20 hours after transfusion, mice were injected IV with freshly prepared hemin and analyzed 4 hours later. (B) Representative dot plots comparing DiI+ PMos (CD45+CD11b+Ly-6G-CD115+Ly-6C−) and CMo (CD45+CD11b+Ly-6G-CD115+Ly-6C+) from WT mice. For the gating strategy used, see supplemental Figure 4B. (C) Frequencies of DiI+ circulating leukocyte subsets in WT mice (n = 8) and CD11a−/− mice (n = 8). (D) Schematic representation of experimental design; Nr4a1-GFP mice received treatment as shown in panel A and were analyzed with immunofluorescence following perfusion with PBS to wash out nonattached circulating blood cells. (E) Representative immunofluorescence images showing the uptake of DiI+ RBCs by GFP+ PMos on the endothelial surface of liver and lung in mice that received DiI-labeled sickle RBCs or control RBCs. CD31/CD144 (endothelial markers, blue), DiI (RBC in red), and GFP (PMo, green). Scale bar, 5 µm. (F) The frequencies of GFP+ PMos with uptake of DiI+ RBC (DiI+ GFP+ PMos) in mice as shown in panel E. (G) Representative IFC images acquired and gated on DiI+ PMos from hemin-treated Nr4a1-GFP reporter mice. Left to right: single-channel BF, GFP (representing PMos in green), DiI (representing sickle RBC material in red), CD45 (representing surface PMos in purple), and merged images showing DiI+ materials within GFP+CD45+ PMos. (H) Histogram depicting degree of DiI+ material internalized within GFP+ PMos with internalization score (<0 represents DiI+ material attached to the surface of PMo; >0 represents DiI+ material internalized by PMos). Percentage of cells with an internalization score >0 is indicated. Data represent mean ± SEM; means were compared using a 2-tailed Student t test. **P < .01; ***P < .001.
To examine whether uptake of sickle RBCs by PMos required attachment to the ECs, DiI-labeled mouse sickle RBCs were transfused into heme-treated Nr4a1-GFP mice, which express GFP in their circulating PMos, followed by perfusion to remove non-EC–attached cells. Using confocal microscopy, we found that GFP+ PMos uptake more DiI+ sickle RBCs than DiI+ control RBCs on the endothelial surface of the vasculature of the lung and liver of perfused recipients, confirming uptake of EC-attached sickle RBCs by PMos (43.3% ± 4.9% vs 7.6% ± 0.7%, P < .001; Figure 3D-F). IFC also revealed that 85% of DiI-labeled sickle RBC materials in DiI+GFP+ PMos were internalized by PMo in Nr4a1-GFP reporter mice (Figure 3G-H). Altogether, these in vivo data further support the in vitro studies that PMos scavenge EC-attached sickle RBCs.
Consequence of sickle RBC uptake by PMos
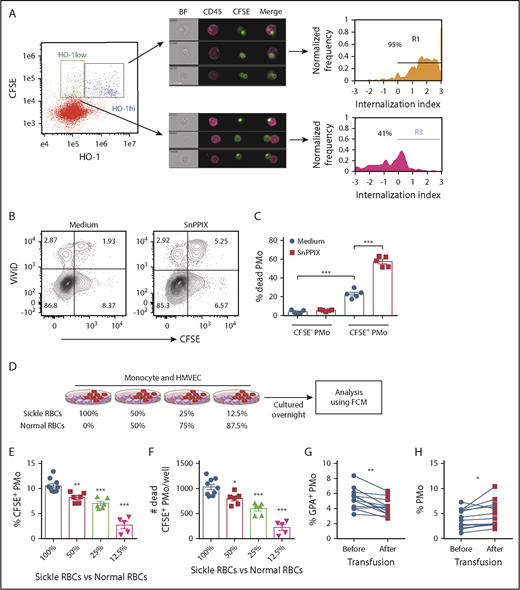
We next examined HO-1 expression levels in erythrophagocytosed PMos and their impact on PMo survival. We found that in cocultures of HD PMos, CFSE-labeled sickle RBCs, and HMVECs, CFSE+ PMos could be divided into 2 populations based on HO-1 expression: CFSE+HO-1hi PMos and CFSE+HO-1low PMos (Figure 4A). The extent of RBC material internalized within the CFSE+HO-1hi PMo subset was higher than that of CFSE+HO-1low PMos (Figure 4A; 95% vs 41%), suggesting that phagocytosis of sickle RBCs induces more HO-1 expression. Interestingly, as compared with CFSE− PMos, CFSE+ PMos had reduced viability (percentage [%] of dead cell marker ViViD+ cells: 4.1% ± 0.8% vs 23.1% ± 2%, P < .001; Figure 4B-C) and increased expression of phosphatidylserine (PS) (% annexin V+ cells: 5.3% ± 0.5% vs 8.9% ± 0.6%, P < .01; supplemental Figure 2A-B). These data suggest that sickle RBC uptake by PMos induces cell death despite HO-1 upregulation. To test the cytoprotective role of HO-1 in PMos during uptake, HO-1 activity was blocked with SnPPIX. We found increased frequencies of dead CFSE+ PMos (ViViD+ cell%: 57.6% ± 2.5%, P < .001; Figure 4B-C) and annexin V+CFSE+ PMos after blockade (annexin V+ cell%: 18.3% ± 0.8%, P < .001; supplemental Figure 2A-B). These data suggest that uptake of sickle RBCs by PMos can lead to apoptosis and that upregulation of HO-1 acts as a cryoprotective mechanism to counteract cell death.
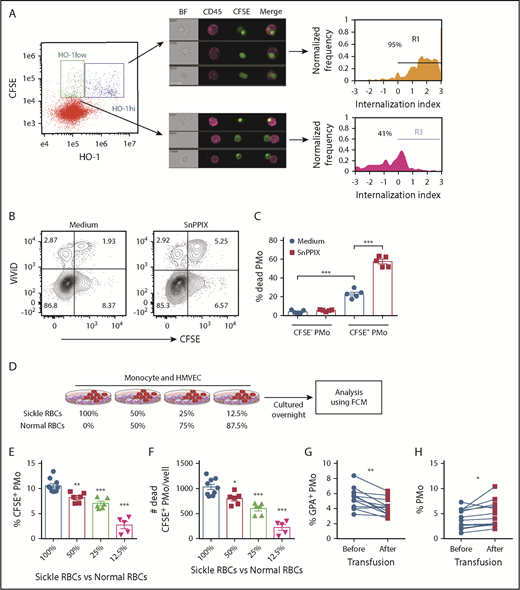
Survival of PMos in SCD. (A) Representative IFC dot plot showing HD PMos gated on CFSE+ HO-1hi and CFSE+ HO-1low subpopulation in cocultures of monocyte/HMVEC/sickle RBCs. CFSE+HO-1hi PMos and CFSE+ HO-1low PMos were further analyzed and shown by representative IFC images and internalization index depicted by histograms. Left to right: single-channel BF, CD45 (representing PMo in purple), CFSE (representing RBC material), and merged images showing the position of CFSE+ materials with CD45+ cells. The internalization score was defined as in Figure 1D. (B) Representative dot plots comparing the frequencies of dead cells (ViViD+ cells) in PMos cultured with CFSE-labeled sickle RBCs and HMVECs in the presence of the HO-1 activity blocker SnPPIX or control (culture media). (C) The frequencies of dead PMos in HD PMo subpopulations of CFSE+ PMos (ie, RBC phagocytosed PMos) and CFSE− PMos (ie, no RBC uptake) are shown as in panel B (n = 5). (D) Schematic representation of experimental design. CFSE-labeled RBCs were mixed with HD RBCs to mimic transfusion in patients with SCD and then cultured with HD monocytes and HMVECs. The uptake of CFSE+ RBCs by PMos was analyzed by FCM. Frequencies of CFSE+ PMos from HD (n = 5-10; E) and absolute number of dead CFSE+ PMos (n = 5-10; F) in the coculture as defined in panel D. (G) Frequencies of GPA+ circulating PMos (G) and percentage of PMos within total monocytes in patients with SCD (n = 12; H) before or after transfusion. Data represent mean ± SEM; means were compared using a 2-tailed Student t test. *P < .05; **P < .01; ***P < .001.
Survival of PMos in SCD. (A) Representative IFC dot plot showing HD PMos gated on CFSE+ HO-1hi and CFSE+ HO-1low subpopulation in cocultures of monocyte/HMVEC/sickle RBCs. CFSE+HO-1hi PMos and CFSE+ HO-1low PMos were further analyzed and shown by representative IFC images and internalization index depicted by histograms. Left to right: single-channel BF, CD45 (representing PMo in purple), CFSE (representing RBC material), and merged images showing the position of CFSE+ materials with CD45+ cells. The internalization score was defined as in Figure 1D. (B) Representative dot plots comparing the frequencies of dead cells (ViViD+ cells) in PMos cultured with CFSE-labeled sickle RBCs and HMVECs in the presence of the HO-1 activity blocker SnPPIX or control (culture media). (C) The frequencies of dead PMos in HD PMo subpopulations of CFSE+ PMos (ie, RBC phagocytosed PMos) and CFSE− PMos (ie, no RBC uptake) are shown as in panel B (n = 5). (D) Schematic representation of experimental design. CFSE-labeled RBCs were mixed with HD RBCs to mimic transfusion in patients with SCD and then cultured with HD monocytes and HMVECs. The uptake of CFSE+ RBCs by PMos was analyzed by FCM. Frequencies of CFSE+ PMos from HD (n = 5-10; E) and absolute number of dead CFSE+ PMos (n = 5-10; F) in the coculture as defined in panel D. (G) Frequencies of GPA+ circulating PMos (G) and percentage of PMos within total monocytes in patients with SCD (n = 12; H) before or after transfusion. Data represent mean ± SEM; means were compared using a 2-tailed Student t test. *P < .05; **P < .01; ***P < .001.
To further examine the consequences of sickle RBC uptake, we cultured monocytes and HMVECs with a serial doubling of CFSE-labeled sickle RBCs from 0.25 million to 4 million per well. The frequencies of CFSE+ PMos increased from 3.7% ± 0.3% to 23.5% ± 1% with increasing numbers of sickle RBCs in culture (P < .001; supplemental Figure 2C). Increased frequencies of CFSE+ PMo dead cells were evident with the highest 2 doses of sickle RBCs tested (P < .05; supplemental Figure 2D), suggesting that increased uptake of sickle RBCs affects PMo survival. By maintaining the same total numbers using a mixture of sickle and HD RBCs, we next altered the ratios of sickle RBCs and HD RBCs in the cocultures to mimic transfusions. Similar to data with sickle RBCs alone, we found reduced frequency of CFSE+ PMos with decreasing ratio of sickle RBCs to HD RBCs (P < .05; Figure 4D-E). The number of dead CFSE+ PMos also decreased correspondingly (Figure 4F). Consistent with the in vitro data, the frequency of GPA+ PMos (within total PMos) was statistically lower in patients with SCD after transfusion compared with before transfusion (P < .01; Figure 4G; supplemental Table 3). In addition, the frequency of PMos (in total monocytes) was increased after transfusion compared with before transfusion (P < .05; Figure 4H). In sickle mice, transfusion with (2 U) WT RBCs, which improved RBC counts and lowered reticulocyte numbers, resulted in an increase in the frequency of PMos, but not CMos, and led to a decrease in the frequencies of Ter-119+ PMos and annexin V+ PMos (P < .05; supplemental Figure 2E-J). Altogether, these data suggest that transfusions decrease sickle RBC uptake by PMos, resulting in lower PMo consumption.
Manipulation of PMo numbers in sickle mice
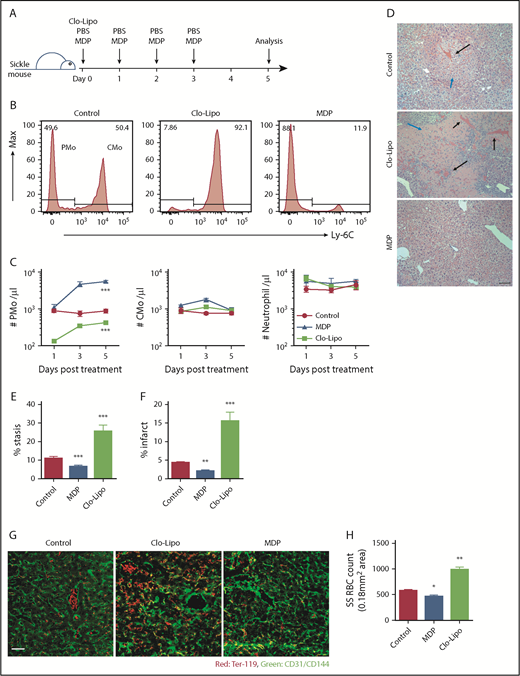
Townes sickle mice develop spontaneous, unprovoked vaso-occlusion.40 To confirm the role of PMos in vascular stasis, we manipulated PMo mass in Townes sickle mice using low-dose Clo-Lipo to selectively deplete PMos35,36 or MDP to induce PMos.34 Using the former treatment, 85% of circulating PMos were depleted at day 1 post-Clo-Lipo injection (10% of normal dose), and only half of the pretreatment PMo numbers were recovered by day 5 (P < .001; Figure 5A-C). In contrast, treatment with MDP in SCD mice increased the circulating PMo numbers by sixfold on day 3, and these numbers were sustained until day 5 (P < .001; Figure 5B-C). Except for an initial twofold increase in CMo numbers on day 3 in MDP-treated mice, no significant change in circulating CMo or neutrophil numbers was detected with either Clo-Lipo or MDP treatment at any time point (Figure 5C). In addition, with this low dose of Clo-Lipo injection, no change in liver (F4/80+) macrophage numbers was detected (supplemental Figure 3A), consistent with the selective depletion of PMos, but not macrophages, as previously reported.35 Histological (H&E) analysis of liver vasculature revealed increased vascular stasis (by over twofold) and infarcts around blood vessels (more than threefold) in mice treated with Clo-Lipo compared with control mice (P < .001); in contrast, mice treated with MDP had fewer (almost half) stasis and infarcts than controls (P < .01; Figure 5D-F). To quantify the number of sickle RBCs trapped in blood vessels, we performed immunofluorescence on liver sections of perfused sickle mice. We found twofold higher numbers of endothelial-attached sickle RBCs in Clo-Lipo–treated mice compared with control (PBS-treated) sickle mice (P < .01; Figure 5G-H). In contrast, mice that had received MDP showed a 20% reduction in the numbers of endothelial-attached sickle RBCs compared with PBS-treated controls (P < .05; Figure 5G-H). The effect of MDP in increasing circulating PMo numbers and decreasing endothelial-attached sickle RBCs, stasis, and infarcts was dose dependent (P < .01; supplemental Figure 3B-G). Altogether, these data demonstrate that PMos guard against vaso-occlusion in a sickle mouse model and that manipulation of PMo numbers has the potential to reduce endothelium-trapped sickle RBCs.
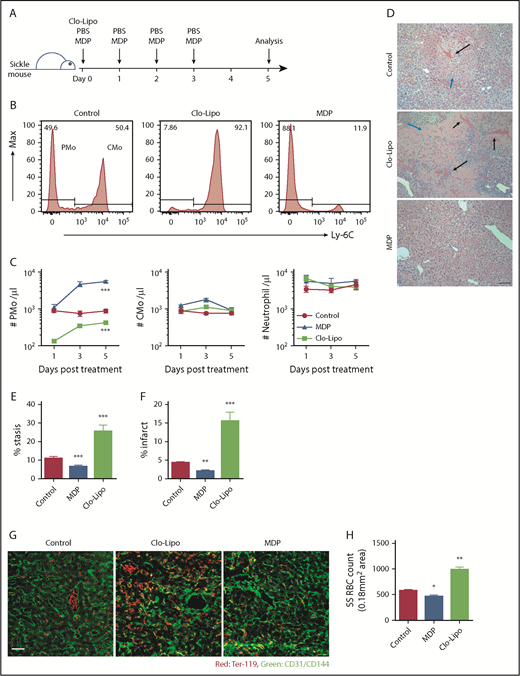
Detection of vaso-occlusion in Townes sickle mice following manipulation of PMo numbers. (A) Experimental design for manipulation of PMo numbers in Townes sickle mice with Clo-Lipo and MDP. Sickle mice (8-12 weeks) were injected IV with MDP daily from day 0 to day 3 or with Clo-Lipo at day 0 or with PBS daily from day 0 to day 3. Mice were sacrificed at day 5 for histological analysis. (B) Representative histograms showing the frequencies of PMos (CD45+CD11b+Ly-6G−CD115+Ly-6C−) and CMos (CD45+CD11b+Ly-6G−CD115+Ly-6C+) in 3 groups of sickle mice as defined in panel A. (C) The absolute number of circulating PMos, CMos, and neutrophils in 3 groups of mice (n = 6-7) as defined in panel A. (D) Representative H&E-stained liver sections in 3 groups of mice (scale bar, 200 µm). Black arrows indicate RBC stasis within blood vessels. Blue arrows indicate infarct. (E) Frequencies of blocked blood vessels (stasis) in total blood vessels in liver sections in 3 groups of mice (n = 6 in each group). (F) Frequencies of area of necrosis (infarct) in liver sections in 3 groups of mice (n = 6 in each group). (G) Representative immunofluorescence staining of liver sections from 3 groups of mice showing CD31/CD144 (endothelial markers, green) and Ter-119 (RBC marker, red). Scale bar, 50 µm. (H) Enumeration of Ter-119+ RBCs per image (0.18 mm2 area) in liver sections in 3 groups of mice (n = 6-7 in each group) as quantified using ImageJ software. Data represent mean ± SEM. Means in panel C were compared using 2-way ANOVA, and means in panel F were compared using a 2-tailed Student t test. *P < .05; **P < .01; ***P < .001.
Detection of vaso-occlusion in Townes sickle mice following manipulation of PMo numbers. (A) Experimental design for manipulation of PMo numbers in Townes sickle mice with Clo-Lipo and MDP. Sickle mice (8-12 weeks) were injected IV with MDP daily from day 0 to day 3 or with Clo-Lipo at day 0 or with PBS daily from day 0 to day 3. Mice were sacrificed at day 5 for histological analysis. (B) Representative histograms showing the frequencies of PMos (CD45+CD11b+Ly-6G−CD115+Ly-6C−) and CMos (CD45+CD11b+Ly-6G−CD115+Ly-6C+) in 3 groups of sickle mice as defined in panel A. (C) The absolute number of circulating PMos, CMos, and neutrophils in 3 groups of mice (n = 6-7) as defined in panel A. (D) Representative H&E-stained liver sections in 3 groups of mice (scale bar, 200 µm). Black arrows indicate RBC stasis within blood vessels. Blue arrows indicate infarct. (E) Frequencies of blocked blood vessels (stasis) in total blood vessels in liver sections in 3 groups of mice (n = 6 in each group). (F) Frequencies of area of necrosis (infarct) in liver sections in 3 groups of mice (n = 6 in each group). (G) Representative immunofluorescence staining of liver sections from 3 groups of mice showing CD31/CD144 (endothelial markers, green) and Ter-119 (RBC marker, red). Scale bar, 50 µm. (H) Enumeration of Ter-119+ RBCs per image (0.18 mm2 area) in liver sections in 3 groups of mice (n = 6-7 in each group) as quantified using ImageJ software. Data represent mean ± SEM. Means in panel C were compared using 2-way ANOVA, and means in panel F were compared using a 2-tailed Student t test. *P < .05; **P < .01; ***P < .001.
Discussion
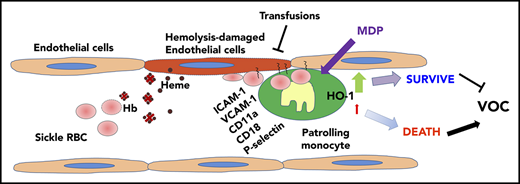
Painful VOC is a hallmark of SCD resulting from adhesive interactions of sickle RBCs and other blood components with the underlying vasculature. In a previous study, we found that PMos protect the endothelium against hemolysis-induced damage.22 Here, we have identified circulating PMos as playing a key role in scavenging endothelium-adherent sickle RBCs and inhibiting vaso-occlusion in the Townes mouse model of SCD. We detected RBC (GPA+)-engulfed material in circulating PMos of patients with SCD, and their frequency was further increased during acute crisis. We demonstrated that PMos uptake RBC endothelial-attached sickle, but not control, RBCs and that engulfment occurs through interactions involving mainly integrins. HO-1 upregulation in PMos counteracts the cytotoxic effects of engulfed RBC breakdown products. Sickle mice depleted of PMos were susceptible to vascular stasis and tissue damage, whereas increasing PMo numbers reduced the numbers of endothelial-attached sickle RBCs and tissue injury. Altogether, these data demonstrate a novel mechanism for removal of endothelial-attached sickle RBCs mediated by PMos that can protect against VOC pathogenesis, opening up the possibility that PMos can be manipulated to clear sickle RBCs that are already attached and resistant to removal from the vasculature.8 Combination therapies using agents that selectively increase PMo sickle RBC phagocytic activity, together with existing/upcoming treatments, may hold promise for a more effective prevention of VOC.
In this study, we demonstrated that circulating PMos uptake sickle RBCs more effectively than other peripheral mononuclear cells in SCD (Figure 1). Surprisingly, PMos did not directly phagocytose sickle RBCs. Instead, PMos engulfed sickle RBCs only in the presence of ECs (Figure 2), and PMo RBC uptake only occurred with sickle RBCs and not HD RBCs. These data are consistent with the model where sickle, but not HD, RBCs adhere to the vascular endothelium.7,41-47 Interestingly, once deoxygenated, sickle RBCs require very large forces to become detached from ECs.48 Circulating PMo as endothelial scavengers possessing potent phagocytotic activity31,33 are thus uniquely poised to remove adherent sickle RBCs. Uptake of sickle RBCs by PMos results in upregulation of HO-1 to detoxify heme/hemoglobin derived from engulfed RBCs. Indeed, PMos express the highest levels of HO-1 compared with other circulating leukocytes in SCD.22,23 Upon uptake of EC-adherent sickle RBCs, we posit that PMos return to the circulation similar to the previously reported microparticle-phagocytosed PMos,32 thus enabling their detection as RBC (GPA+)–engulfed PMos in the circulation of patients with SCD. In this model, we further hypothesize that hemolysis (through free hemoglobin or labile heme) or inflammation (eg, TNF-α) that contributes to EC activation (higher ICAM-1, VCAM-1, P-selectin, and E-selectin levels) leads to increased numbers of attached sickle RBCs,49-52 resulting in additional PMos that engulf the adherent cells (Figure 2C). Similar to the in vitro system, PMos captured and engulfed endothelium-trapped sickle RBCs in vivo (Figure 3). It remains to be determined whether PMos can also uptake other EC-attached blood components such as platelets, sickle RBC–derived microparticles, or even neutrophils known to induce sickle VOC.12,53,54
We also examined the molecules involved in PMo engulfment of EC-attached sickle RBCs using antibody blockade experiments. RBC uptake was inhibited with antibodies against integrins CD11a/CD18 (lymphocyte function–associated antigen 1 [LFA-1]), consistent with the role of LFA-1 in PMo patrolling function.32,37 Furthermore, antibodies against ICAM-1 and VCAM-1, which act as ligands for LFA-1,32 also blocked sickle RBC uptake (Figure 2E). These data demonstrate that the interaction between PMo and EC through LFA-1–ICAM-1/VCAM-1 are important for sickle RBC uptake by PMo. It should be noted that VCAM-1 on ECs is required for binding to sickle RBCs,55 and therefore, anti-VCAM-1 likely inhibited sickle RBC–EC interactions, reducing the numbers of EC-attached sickle RBCs available to PMos for uptake. Antibodies to P-selectin, which is expressed by ECs and plays a key role in sickle RBC attachment to the endothelium,56 also inhibited sickle RBC uptake by PMos. Indeed, a monoclonal antibody to P-selectin (crizanlizumab),57 by inhibiting adherence of sickle RBCs to ECs, and a pan-selectin inhibitor (rivipansel, GMI-1070),58 have shown efficacy in reducing pain crisis in SCD trials. In our studies, we propose that anti-P-selectin blocks sickle RBC–EC interactions, but not monocyte–EC interactions, since antibodies to PSGL-1, which is the P-selectin receptor and is expressed on monocytes, did not inhibit uptake. Blocking Fc receptors (CD32, CD64, or Fc α/μ receptor), which are key players in IgG-mediated erythrophagocytosis, had little effect on sickle RBC uptake by PMos. Consistent with this, extremely low IgG levels were present on both sickle and HD RBCs. In addition, uptake by PMos was unperturbed in IgG-depleted culture conditions (supplemental Figure 1F-H). Nevertheless, the anti-CD16 antibody (3G8), which has been shown to trigger a signaling event in natural killer cells with functional consequences (lower ADCC),59 led to a reduction in PMo engulfment of sickle RBCs. We speculate that this blocking antibody may impact PMo phagocytotic functions, but this needs to be further confirmed.
PMo numbers are lower in patients with SCD than in HDs and even lower in patients experiencing acute pain crisis (Figure 1E). We propose that phagocytosis of sickle RBCs leads to increased cell death. Indeed, we found increased numbers of dead PMos following uptake of sickle RBCs (Figure 4). A similar phenomenon has also been reported in red pulp macrophages following phagocytosis of RBCs in non-SCD mice.60 Additional mechanisms responsible for lower PMo numbers may include clearance of apoptotic/necrotic PMos by macrophages in spleen/liver/bone marrow similar to removal of other senescent/necrotic/apoptotic circulating blood cells.61,62 During acute crisis, patients present with an inflammatory cytokine profile,63-65 which may affect PMo survival. However, in severe inflammatory conditions such as sepsis, frequencies of PMo and monocyte numbers are increased (not decreased).66,67 Inflammatory cytokines therefore may not be responsible for decrease in PMo during crisis, although this has not been tested directly.
HO-1 activity blockade during PMo phagocytosis increased PMo death (Figure 4B-C) but interestingly had no effect on sickle RBC uptake (Figure 2E). These data suggest that once phagocytosed, engulfed sickle RBCs and/or its derived hemoglobin/heme60 can impair PMo viability and that HO-1 upregulation can counteract this effect. Although not yet studied, iron chelation may improve PMo survival, since we suspect that RBC uptake by PMos may lead to ferroptosis, an iron-dependent form of cell death due to erythrophagocytosis.68 In support of a role for HO-1 in protection against SCD complications, inhibition of HO-1 activity exacerbated vaso-occlusion in SCD.69 It should be noted that hemolysis-endothelial-expressed HO-1 and its products may also confer protection against EC activation, thereby inhibiting white blood cell interactions with the vasculature.52,70 In myeloid cells, targeted HO-1 deletion induced inflammation during reperfusion injury in a non-SCD setting,71 while HO-1 overexpression inhibited lipopolysaccharide-stimulated TNF-α expression in human monocytes.72 We have also found lower expression of TNF-α and IL-6 cytokines in HO-1hi as compared with HO-1low SCD PMos.22 Thus, the anti-inflammatory effect of HO-1 induction in PMos may be an additional mechanism of protection.
By changing the ratio of sickle RBCs to HD RBCs in our in vitro culture model of transfusion as well as comparing post- vs pretransfusion in patients with SCD, we were able to demonstrate a decrease in the number of sickle RBC–engulfed PMos and an increase in PMo viability following transfusion. The ex vivo measurements were performed within 4 hours of transfusion, and hence, we expect that the effects seen, which were small, albeit significant (Figure 4G-H), would be more apparent after a few days after transfusion. Consistent with this, transfusions into Townes sickle mice decreased the number of RBC-engulfed PMos and reduced PMo consumption at later time points (day 4). We posit that the resulting increase in PMos may in part explain why chronic transfusions protect against the severity of VOC in patients with recurrent pain crisis.
A striking observation of our study was that manipulating PMo numbers altered vascular stasis in Townes sickle mice, a preclinical model of SCD. We postulate that an increased number of PMos is important to decrease endothelium-attached sickle RBCs in SCD. We had previously shown that transfusion of sickle RBCs to mice lacking PMos (Nr4a1 KO mice) results in increased sickle RBCs trapped on the vascular endothelium.22 In this study, we were able to directly manipulate PMo numbers in SCD mice and show that targeted depletion of PMos increased stasis and infarcts, whereas increasing PMo mass using MDP reduced stasis and tissue damage (Figure 5). MDP treatment was shown to increase anti-inflammatory cytokines (IL-10 and TGF-β) in peritoneal lavages in a peritonitis model and decreased inflammatory cytokines (TNF-α and IL-6) in a model of lipopolysaccharide-induced inflammation,34 indicating that it can also alter the inflammatory milieu. Thus, MDP, which is a ligand for NOD2 with the ability to activate other cells,73 can paradoxically reduce inflammation and increase PMo numbers. To circumvent the potential non-PMo effects, future studies are aimed at optimizing MDP delivery by packaging MDP into liposomes that directly target monocytes rather than other cells.74
In summary, our data demonstrate for the first time that PMos scavenge and remove endothelium-trapped sickle RBCs, further supporting a key role of PMos as ‘‘housekeepers’’ of the SCD vasculature. We postulate that deficiency in PMo numbers or PMo HO-1 activity hampers their endothelial scavenger function in SCD, which in turn results in the precipitation of VOC. Furthermore, the data suggest that strategies to increase PMo numbers and functions, such as using MDP and/or transfusions to promote PMo survival, may represent a therapeutic approach against VOC.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kathy Tang (New York Blood Center) for mouse maintenance, Mihaela Barbu-Stevanovic (New York Blood Center) for cell sorting, and services provided by the Memorial Sloan Kettering Pathology Department for animal tissue sections and H&E staining.
This study was supported by National Heart, Lung, and Blood Institute, National Institutes of Health grants R01HL121415 (K.Y.) and R01HL130139 (K.Y.).
Authorship
Contribution: Y.L. conceived the study and performed and analyzed all the experiments; W.B. assisted with sickle mouse studies; H.Z., A.M., X.A., and P.S. assisted with experimental procedures and analysis; D.M. and S.T.C. were involved with all aspects of selection, recruitment, and provision of blood samples from patients and controls; K.Y. was responsible for experimental design, manuscript writing, and project supervision; and Y.L. and K.Y. wrote the manuscript with consultation and contribution from all coauthors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Karina Yazdanbakhsh, Laboratory of Complement Biology, New York Blood Center, 310 E 67th St, New York, NY 10065; e-mail: kyazdanbakhsh@nybc.org.

![Figure 2. PMo uptake sickle RBCs when cocultured with HMVEC. (A) Schematic representation of experimental design. HMVECs were pretreated with hemin or PBS for 2 hours and subjected to 2 washes before the addition of purified total monocytes and CFSE-labeled RBCs. Some monocytes and labeled RBCs were cultured in the absence of HMVECs. After overnight cultures, the uptake of CFSE+ RBCs by monocyte subsets was analyzed by FCM and IFC. (B) Representative dot plots comparing CFSE+ PMos and CFSE+ CMos from cultures shown in panel A. Frequencies of CFSE+ monocyte subsets from HDs (n = 3-12; C) and patients with SCD (n = 6-12; D). (E) HMVECs were preincubated with individual blocking antibodies (anti-ICAM-1, anti-VCAM-1, anti-CD11a, anti-CD11b, anti-P-selectin, anti-PSGL-1, anti-CD11c, anti-CD18, anti-CD31, anti-CD16, anti-CD32, anti-CD64, anti-Fc α/µ receptor, or isotype matched control [mouse IgG] antibodies) or annexin V or tin protoporphyrin IX (SnPPIX) for 30 minutes before the addition of HD monocytes and CFSE-labeled sickle RBCs. It should be noted that it takes ∼10 to 15 minutes for monocytes and RBCs to settle on the HMVEC. Frequencies of CFSE+ PMos were then analyzed (n = 4-12). Data represent mean ± SEM; means were compared using a 2-tailed Student t test. *P < .05; ***P < .001. Ab, antibody.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/134/7/10.1182_blood.2019000172/3/m_bloodbld2019000172f2.png?Expires=1767861936&Signature=qrNF7Vz8ytxD47rKkzW79skuiA5IqW98zW4rFoKUx-fFd6fl3hYHzOk6q9~ci4KBSrMjfnP70Y359rsFthEdlsAEixFoC1d-oJPfvBRaj9CWvUofJwtxgGmhZ-rHNMldhApEXlg0BVwhpm594hmGXx~hSIXlX8nmhAe7eKE9-UBG-bN-wnZeGWsLoTyZNUVfFyzcVKM1MLxd~SLRVfbZup6moGIeAO-zr5-oHdu9vcUmgvAEMP~W6wis4OqdSIoJOhczjTp-s1ijRe3Dr-LUNTh9vUzLPqCgrWAdHFvCJZwDSzu0GXlFd7EHbuO2n8d~6l~OiTCQXdDS8LjYMmd45g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)