

Introduction:
Core binding factor acute myeloid leukemia (CBF-AML) is one of the commonest subtypes of AML characterized presence of t(8;21)(q22;q22) or inv(16)(p13q22)/t(16;16)(p13;q22). It is characterised by a high frequency of somatic mutations especially in RAS and tyrosine kinase signalling pathways. Here we investigated the feasibility of improving risk prediction of CBF-AML using machine learning algorithms.
Methods:
We developed a next generation sequencing panel that targeted 50 genes implicated in the pathogenesis of myeloid malignancies using single molecule molecular inversion probes. This panel was used to sequence 106 patients of CBF-AML accrued over a six year period (March 2012 - December 2018) treated with conventional "3 + 7" chemotherapy. Post data analysis, we devised a supervised machine learning (ML) approach for identification of mutations most likely to predict for favorable outcome in CBF-AML. We included somatic mutations in genes occurring in CBF-AML at a frequency of >5%. A total of 11 variables were included for feature selection to predict for favorable outcome (including mutations in ASXL2, CSF3R,FLT3, KIT, NF1, NRAS, RAD21, TET2 and WT1 genes as well as mutation burden). Approaches for supervised ML were naïve bayes, generalized linear model, logistic regression, deep learning and random forest methods. Based on the ML results top 6 selected variables were allotted an individual score. A final score for that case was devised as a sum total of the individual scores. These sum were used to generate a genetic risk for a patient. Overall survival (OS) was calculated from date of diagnosis to time of last follow up or death. Relapse free survival (RFS) was calculated from date of CR till time to relapse or death or last follow up if in CR. Results of the genetic risk were analyzed for their impact on OS and RFS using log rank test. Multivariate analysis was performed using cox proportional hazards regression model.
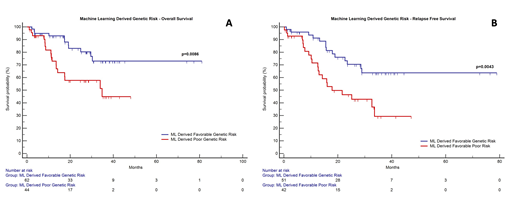
Results: The median follow up of the cohort was 27.6 months. A total of 181 somatic mutations were identified in this subset of AML with 86.7% harbouring at least one somatic mutation (median = 2). Based on ML data, a genetic score was formulated that incorporated mutations in RAD21, FLT3, KIT D816, ASXL2, NRAS genes as well as high mutation burden (≥2) into two genetic risk classes (favorable risk and poor ML derived genetic genetic risk). Patients classified as poor genetic risk had a significantly lower OS [median OS: 34.8 months; 95% confidence interval (CI) (14.2-34.8); p=0.0086] and RFS [median RFS: 17.9 months; 95%CI (12.7-33.6); p=0.0043] as compared to patients with favorable genetic risk (median OS and RFS not reached). These results can be seen in Figure 1. On multivariate analysis poor genetic risk was the most important independent risk factor that predicted for inferior OS [hazard ratio(HR), 2.7; 95% CI 1.3 to 5.7] and RFS (HR, 2.6; 95% CI:1.3 to 5.1).
Conclusions
In a proof of concept, we describe a novel ML derived genomics scoring model that provides a mechanism to risk stratify CBF-AML, a seemingly homogeneous disease entity. This study, to the best of our knowledge represents a novel application of ML to CBF mutated AML. Our data indicates that this scoring system will be useful in identifying CBF mutated AML patients who are at higher risk of relapse and distinguishes them from patients who are truly good risk.
No relevant conflicts of interest to declare.

