

Introduction: High dose therapy and autologous stem cell transplant (ASCT) remain standard first line therapy for young fit patients with multiple myeloma (MM). Beyond cytoreduction, ASCT may also alter the bone marrow (BM) immune environment to augment anti-tumour immune responses. A more precise understanding of the immune microenvironment post ASCT and its relationship with clinical outcomes may identify biomarkers to refine prognostication and provide opportunity for therapeutic intervention.
Aim: We aimed to define T cell subsets and expression of co-activating and co-inhibitory proteins in the BM early post ASCT, and explore associations with clinical outcomes.
Methods: BM samples were obtained at 100 days (D100) post ASCT from 61 MM patients, and 15 healthy donors (HD). BM mononuclear cells (MNCs) were stained for surface (CD3, CD4, CD8, PD-1, LAG-3, and ICOS) and intracellular markers (GzmB, Ki-67, CTLA-4, and FoxP3). Data were acquired on a BD LSR Fortessa and analysed using FlowJo. Progression free survival (PFS) was defined from ASCT as per IMWG criteria, and patient groups were compared using Mann-Whitney U test. Adverse risk genetics was defined as t(4;14), t(14;16) or del(17p).
Results: Median age of patients at ASCT was 58yrs (36-71), 70.5% were male. Median follow up was 17 months (3-54). At diagnosis, 36.1% were ISS stage I, 8.2% had extramedullary disease, and 14.7% had adverse risk genetics. As induction therapy, 96.7% received a proteasome inhibitor, 45.9% an immunomodulatory drug, 54.1% both, and 21.3% received post ASCT maintenance/consolidation. Median PFS was 24 months. At D100, 24.6% had achieved CR, 47.5% VGPR, 21.3% PR, 4.9% SD and none had PD. Improved PFS was found in patients with deeper response (CR/VGPR vs. rest, p=0.003), and with ISS stage I vs II/III (p=0.013). There was a trend for improved PFS with standard risk genetics (p=0.088 cf high risk).
MM BM contained lower frequencies of CD3 and CD4 T cells compared to HD (CD3, 28% of live MNCs, vs 41.2%, p=0.012; CD4, 4.1% vs 14.0%, p<0.0001), while CD8 frequency was similar. The frequency of regulatory T cells (CD4+FoxP3+, Tregs) was higher in MM patients compared to HD (0.23% of live MNCs vs 0.07%, p=0.02; 4.2% of CD4+ vs 1.2%, p<0.0001). Thus CD4 effector (CD4+FoxP3-, CD4eff):Treg ratio was lower in MM patients (16.8 vs 140.2, p<0.00001). There was no difference in CD8:Treg ratio.
Immune checkpoint proteins were highly expressed on BM T cell subsets in post ASCT compared to HD. CD4eff in MM patients expressed higher frequency of PD-1, LAG-3, ICOS, CTLA-4, GzmB, and Ki-67 (p<0.0001, p=0.0016, p=0.0018, p=0.003, p<0.0001, and p<0.0001, respectively). CD8 T cells in MM patients expressed higher frequency of LAG-3, GzmB and Ki-67 (p=0.0003, p<0.0001, and p<0.0001 respectively). We observed higher frequencies of ICOS and CTLA-4 on Tregs in MM patients (vs HD p=0.0025 for both) indicating Treg activation and enhanced suppressive function. There were considerable correlations between the frequency of LAG-3 on different T cell subsets; (CD4eff LAG-3+ vs CD8 LAG-3+ r=0.82, p<0.0001; CD4eff LAG-3+ vs Tregs LAG-3+, r=0.56, p<0.0001; Tregs LAG-3 vs CD8 LAG-3, r=0.5, P<0.0001).
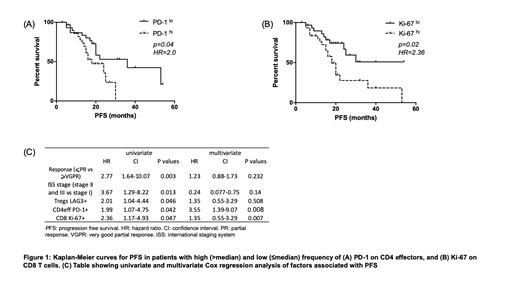
Clinical outcomes were not influenced by T cell subsets (CD3, CD4eff, CD8, and Treg) but there was an inverse association with co-inhibitory receptor expression. High frequency (>median) of LAG-3 on CD4eff and CD8, and Tregs was associated with shorter PFS (median 18mo vs 53, p=0.0028; 20mo vs 53, p=0.0057; 20mo vs 53, p=0.046 respectively). Further, patients with high frequency of PD-1 on CD4eff and Ki-67 on CD8 T cells had shorter PFS (median 18mo vs 36, p=0.042 and 18mo vs unreached, p=0.019 respectively). A multivariate Cox regression model was built to include ISS stage, depth of response at D100, and the immune phenotypes identified to correlate with PFS. In this model, high frequency of PD-1 on CD4eff retained independent prognostic value, along with ISS stage and CD8 Ki67+ T cells.
Conclusion: We present data indicating that high frequency of PD-1+ CD4eff and Ki67+ CD8 T cells post ASCT associate with shorter PFS in MM patients. Pending confirmation in larger cohorts, these data reveal potential immune biomarkers for poor outcomes, and prompt deeper phenotypic analysis and mechanistic studies to uncover the immune drivers of sustained remissions post ASCT.
Lee:Autolus Therapeutics: Equity Ownership, Research Funding. Yong:Takeda: Research Funding, Speakers Bureau; Autolus: Consultancy; Amgen: Research Funding, Speakers Bureau; Janssen: Speakers Bureau; Sanofi: Speakers Bureau.

