

INTRODUCTION
Structural variants (SV) include copy number aberrations (CNA) and balanced chromosomal rearrangements. Knowledge of SVs is important for diagnosis and prognosis of myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML), and can alter therapy in some cases. However, high-throughput identification for diagnostic use in a CLIA-certified laboratory is limited by the need for a combination of techniques to detect both CNA and balanced rearrangements. Karyotyping evaluates 20 metaphases from dividing cells. Fluorescence in situ hybridization (FISH) targets known SVs. Microarray is less sensitive and cannot detect balanced rearrangements. While exome sequencing has limited ability to detect medium-large SVs and those adjoining repetitive sequences. Next-generation optical mapping (OM) is a novel technique to identify SVs by de novo assembly of extremely long-read fluorescently labeled DNA sequences against a reference followed by serial imaging. We hypothesize that OM could potentially offer a single platform for identification of all SVs.
METHODS
High molecular weight DNA was isolated from fresh bone marrow mononuclear cells preserved in DMSO. Minimum number of cells required was 1 million. A minimum of 36 ng/dL was required based on initial validation studies using cell lines. Specific sequences across the entire genome were labeled using a direct labelling enzyme (DLE-1). The generated long chromosomal fragments underwent high-throughput high-resolution imaging of long chromosomal fragments resulting in >100X genome coverage on Saphyr system (Bionano Genomics, San Diego). Algorithms to convert images to molecules was followed by de novo assembly of genome maps with reference [hg38]. Chromosomal aberrations were identified by comparing to a reference dataset that includes 200 healthy controls that facilitates detection of all structural abnormalities at a resolution of > 500 bp. Only SVs not identified in the control sample database with recommended variant confidence scores and strong molecule support were considered. Insertions, deletions and duplications larger than 100 kbp were considered. Based on sensitivity studies performed using simulations, the level of detection (LOD) was ~95% for SVs with allele fraction of ~10%. The generated data was compared with results from karyotype, FISH and microarrays (results were blinded during analysis).
RESULTS
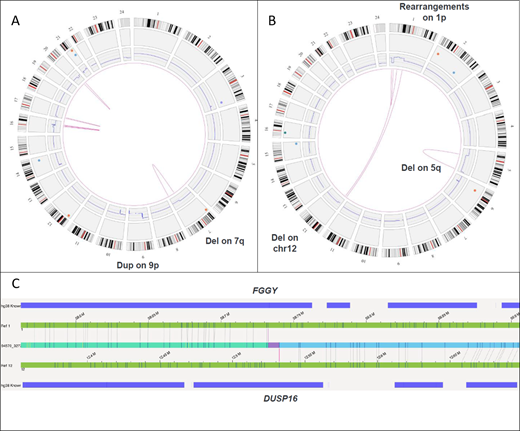
A total of 7 MDS samples with 17 previously known aberrations were selected: 4 with complex karyotype (CK) that included deletions, insertions and translocations [2-way such as t(9;11), 3-way: t(2;20;17)] leading to derivative chromosomes and 3 with normal karyotype (NK). All 7 samples successfully underwent DNA extraction with an average yield of 60 ng/dL. OM identified all SVs detected by karyotype/ array CGH. This included deletions, insertions and translocations. Sixteen of 17 SVs identified by karyotype alone were detected by OM. The only SV missed was der(7)add(7)(p13)add(7)(q32), noted in 3 of 20 (~15% cell fraction) metaphases. With this knowledge, upon re-review, SV was detectable in the raw data at an allele burden of 7.5%, which is below the assay's LOD. For the same reason, this SV was not identified by array CGH (assay detection level ~20%). OM identified multiple additional SVs not detected by conventional techniques. In 4 samples with CK, a total of 6 new SVs were noted. This included del(17p) [25% allele fraction] involving TP53 gene in one patient (pt) [Fig 1A]. Identification of TP53 loss in this MDS pt with concurrent TP53 mutation has important prognostic and therapeutic implications. A novel fusion t(1;12) FGGY-DUSP16 was identified in another pt [Fig 1B,C] along with 2.1 Mb chr(6) deletion and 114 Mb chr(16) duplication. In 3 samples with NK, OM detected additional deletion of chr(19) involving genes: TCF3, GNA11, MAP2K2, SH3GL1, MLLT1 in one pt. No additional SVs were found in remaining 2 NK samples. OM facilitated precise mapping of genomic co-ordinates of SVs, especially with complex rearrangements. The LOD from these samples was estimated at ~10% allele fraction.
CONCLUSIONS
High concordance between OM and conventional techniques provides proof-of-concept for potential use of OM technology as a single-platform for comprehensive assessment of all SVs (CNAs and balanced rearrangements) in MDS at a resolution comparable to standard-of-care assays without the need for cell culture.
Sasaki:Otsuka: Honoraria; Pfizer: Consultancy. Kantarjian:Cyclacel: Research Funding; AbbVie: Honoraria, Research Funding; Immunogen: Research Funding; Novartis: Research Funding; Ariad: Research Funding; Actinium: Honoraria, Membership on an entity's Board of Directors or advisory committees; Daiichi-Sankyo: Research Funding; Amgen: Honoraria, Research Funding; Astex: Research Funding; Agios: Honoraria, Research Funding; Pfizer: Honoraria, Research Funding; Jazz Pharma: Research Funding; BMS: Research Funding; Takeda: Honoraria. Bueso-Ramos:Incyte: Consultancy. Garcia-Manero:Amphivena: Consultancy, Research Funding; Helsinn: Research Funding; Novartis: Research Funding; AbbVie: Research Funding; Celgene: Consultancy, Research Funding; Astex: Consultancy, Research Funding; Onconova: Research Funding; H3 Biomedicine: Research Funding; Merck: Research Funding.

