

Lenalidomide, bortezomib, melphalan and dexamethasone are standard drugs for the treatment of multiple myeloma (MM). Although many patients initially respond to treatment regimens including these drugs, the majority ultimately relapses due to the development of resistance of the MM cells, what may result from acquired genetic alterations.
Here we performed fluorescence in situ hybridization (FISH) and whole exome sequencing (WES) on 16 paired pre-treatment/ progression MM samples followed by functional validation through CRISPR/Cas9-based screens to identify gene mutations that are associated with resistance.
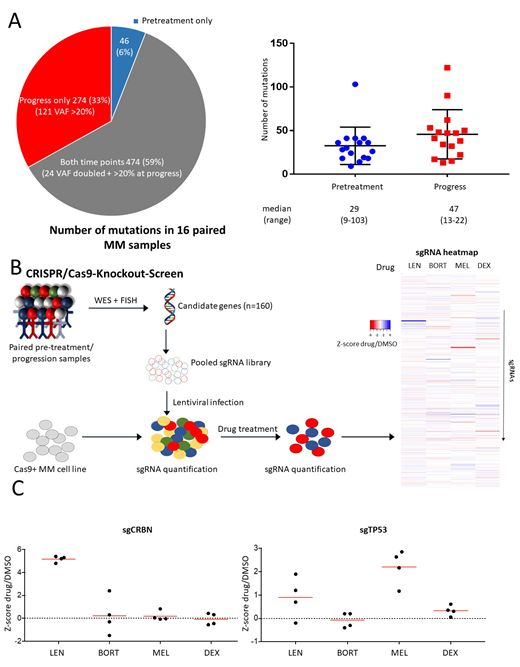
Treatment between two samples consisted of lenalidomide (n=16), bortezomib (n=14), dexamethasone (n=16) and melphalan (n=9). Cytogenetic analyses by FISH revealed that the majority of translocations (7 of 10) and chromosomal gains and deletions (22 of 28) were concordant between pre-treatment and relapse samples. In contrast, gene mutations assessed by WES were highly variable: of the total of 794 identified mutations 6% (n=46) were present only at diagnosis, 59% (n=474) at both time points and 35% (n=274) specifically at relapse with an increase of the median number of mutations from 29 (range 9-103) in pre-treatment to 47 (range 13-110) in progression samples (figure 1A). Recurrent mutations detected pretreatment were in general stable at progression: NRAS (3/3), KRAS (4/4), IGLL5 (3/3) and DIS3 (2/3). Only very few of the newly acquired gene mutations at progression were recurrent: TP53 (n=4), DNAH5 (n=4) and WSCD2 (n=3) while the remaining were non-recurrent.
In order to investigate the functional impact of relapse-specific gene mutations on drug resistance we performed pooled CRISPR-Cas9-based knockout screens (figure 1B). We included 160 gene mutations that fulfilled the following criteria: 1) a variant allele fraction (VAF) of >20% at the time of progression, 2) found exclusively in progression samples or had a more than 2-fold increase in VAF at progression as compared to pre-treatment, 3) predicted to be loss-of-function. In addition, we included genes found recurrently mutated in relapsed MM in previously published studies. Resistance screens were performed in three different MM cell lines (MM1S, NCIH-929, KMS27) with 4 sgRNA per gene in the presence of lenalidomide, dexamethasone, melphalan, bortezomib or DMSO as a control. None of the sgRNAs included in the screen conferred resistance to all four drugs. In contrast, we identified several genes whose inactivation caused resistance to a specific drug. For lenalidomide, the top hits were members of the CRBN-CRL4 E3 ubiquitin ligase, the primary target of IMiDs, including CRBN, CUL4B and DDB1. CRISPR-mediated inactivation of these genes was specifically associated with lenalidomide resistance since sensitivity towards other drugs was not affected. In addition, we found sgRNAs targeting SYT5, a membrane protein involved in Ca2+-dependent exocytosis, to be enriched in lenalidomide-treated cells that so far has not been related to lenalidomide resistance. SgRNAs targeting TP53 were also weakly enriched after lenalidomide treatment in two of the three cell lines but conferred a high level of resistance to melphalan in all three cell lines (figure 1C). Consistently, three of the four TP53 mutations identified by WES were detected in samples obtained after cytotoxic chemotherapy and one after 3 years of treatment with lenalidomide/dexamethasone. Our screens also revealed an increased susceptibility to melphalan by inactivation of ATM, FANCA, BIRC3, and BRCC3, all involved in DNA damage repair. The top sgRNAs causing resistance to dexamethasone were directed against ANKMY2 and BIRC3 in two cell lines (MM1S and NCI-H929). For bortezomib, inactivation of only one gene, TMC2, encoding a transmembrane protein was associated with resistance in two cell lines whereas BIRC3 inactivation provided increased susceptibility to bortezomib.
In conclusion, by combination of comprehensive genetic analyses of tumor samples before and after treatment with functional genetic screens we found mutations that are causally linked with drug-specific resistance and sensitivity. These results may help to personalize therapy in patients with multiple myeloma.
Bohl:Pfizer: Honoraria. Döhner:Celgene, Novartis, Sunesis: Honoraria, Research Funding; AbbVie, Agios, Amgen, Astellas, Astex, Celator, Janssen, Jazz, Seattle Genetics: Consultancy, Honoraria; AROG, Bristol Myers Squibb, Pfizer: Research Funding. Bullinger:Astellas: Honoraria; Bristol-Myers Squibb: Honoraria; Celgene: Honoraria; Daiichi Sankyo: Honoraria; Gilead: Honoraria; Hexal: Honoraria; Janssen: Honoraria; Jazz Pharmaceuticals: Honoraria; Menarini: Honoraria; Novartis: Honoraria; Pfizer: Honoraria; Sanofi: Honoraria; Seattle Genetics: Honoraria; Bayer: Other: Financing of scientific research; Abbvie: Honoraria; Amgen: Honoraria. Krönke:Celgene: Membership on an entity's Board of Directors or advisory committees, Speakers Bureau.

