

Key Points
Adaptive resistance is often associated with TP53 abnormalities or kinase activation, particularly FLT3 internal tandem duplication.
NPM1 mutation is associated with excellent survival prospects and durable molecular remission after venetoclax-based combination therapy.

Abstract
The BCL-2 inhibitor venetoclax combined with hypomethylating agents or low-dose cytarabine represents an important new therapy for older or unfit patients with acute myeloid leukemia (AML). We analyzed 81 patients receiving these venetoclax-based combinations to identify molecular correlates of durable remission, response followed by relapse (adaptive resistance), or refractory disease (primary resistance). High response rates and durable remissions were typically associated with NPM1 or IDH2 mutations, with prolonged molecular remissions prevalent for NPM1 mutations. Primary and adaptive resistance to venetoclax-based combinations was most commonly characterized by acquisition or enrichment of clones activating signaling pathways such as FLT3 or RAS or biallelically perturbing TP53. Single-cell studies highlighted the polyclonal nature of intratumoral resistance mechanisms in some cases. Among cases that were primary refractory, we identified heterogeneous and sometimes divergent interval changes in leukemic clones within a single cycle of therapy, highlighting the dynamic and rapid occurrence of therapeutic selection in AML. In functional studies, FLT3 internal tandem duplication gain or TP53 loss conferred cross-resistance to both venetoclax and cytotoxic-based therapies. Collectively, we highlight molecular determinants of outcome with clinical relevance to patients with AML receiving venetoclax-based combination therapies.
Introduction
Acute myeloid leukemia (AML) is an aggressive hematopoietic malignancy, with disease incidence rising sharply among older patients.1 Treatment options for unfit older patients have historically been limited, with DNA methyltransferase inhibitors (DNMTi’s; ie, azacitidine and decitabine) and low-dose cytarabine (LDAC) providing only modest benefit.2,3 Although higher clinical responses may be achieved with intensive chemotherapy, durability is generally short lived and counterbalanced by higher toxicities.
On 21 November 2018, the US Food and Drug Administration approved the selective BCL-2 inhibitor venetoclax in combination with either DNMTi’s or LDAC in older or unfit patients with AML. In a study of 145 patients ≥60 years, venetoclax in combination with DNMTi’s was associated with complete remission (CR) or CR with incomplete hematologic recovery (CRi) in 67%, with a median overall survival (OS) of 17.5 months.4 In 82 patients ≥60 years receiving venetoclax in combination with LDAC, the CR/CRi rate was 54%, and median OS was 10.1 months.5
Despite these promising responses, primary resistance and clonal evolution leading to adaptive resistance is a recurring theme in AML. Recent studies utilizing single-cell technologies have illustrated the complex and polyclonal nature of resistance to targeted therapeutics including IDH and FLT3 inhibitors.6,7 It is now clear that multiple resistant subclones evolving contemporaneously during therapy can occur in AML and act as a barrier to the long-term success of targeted therapies. We have previously reported molecular correlates of AML response in patients with relapsed or refractory disease treated with venetoclax monotherapy, a setting in which the CR rate was only 19%. Blast reductions were associated with SRSF2/ZRSR2 and IDH1/2 mutations, whereas FLT3 internal tandem duplication (ITD) and PTPN11 variants were linked to primary and adaptive resistance.8 Currently, however, venetoclax is principally used in combination with DNMTi’s or LDAC in previously untreated patients due to the greater activity observed. The objective of this current work, therefore, was to gain insight into clinically relevant genomic factors influencing treatment outcome among patients receiving these frontline venetoclax-based combination therapies.
In summary, we identify FLT3-ITD and other kinase-activating mutations, as well as TP53 aberration, as major determinants of adaptive drug resistance. At the single-cell level, we identified polyclonal emergence of multiple independent kinase activating clones, including FLT3-ITD, FLT3-TKD, FLT3 N676K, and RAS mutations. Functional studies demonstrated resistance to venetoclax alone as well as in cytotoxic drug combinations in the presence of FLT3-ITD gain or TP53 loss. In some cases that were refractory based on standard morphologic assessment, interval assessment revealed differential selection of drug-resistant leukemic clones evolving after a single cycle of treatment, as well as interval reduction of other clones. In contrast, NPM1mut and IDH2mut were associated with high rates of response and durable remissions. In NPM1mut AML, measurable residual disease (MRD) was eliminated in most cases. These findings highlight molecular determinants of long-term response and adaptive resistance to venetoclax-based combination therapy that will be useful for guiding future management.
Methods
Sample acquisition
Samples were collected from patients treated at the Alfred hospital and the University of Texas MD Anderson who had consented to participate in research. The samples were collected as part of routine treatment and were stored and processed through the MD Anderson Molecular Diagnostics Laboratory or Alfred Hospital Tissue Bank. Ethical oversight of the project was through the Human Research Ethics Committee of the Alfred Hospital (Project 450/17), the Walter and Eliza Hall Institute of Medical Research (Project 13/01), the University of Texas MD Anderson Cancer Center, and the Declaration of Helsinki. The use of the sequencing data is subject to a data transfer agreement and is restricted to ethically approved research into blood cell malignancies and cannot be used to assess germline variants.
Clinical details
Patients were treated on 1 of 2 trials4,5 and received either venetoclax with a DNMTi (azacitidine or decitabine) or LDAC. Cytogenetic risk was classified in both studies in accordance with the revised Medical Research Council classification.9 Molecular profiling was performed using several targeted exon panels (detailed in supplemental Methods, available at the Blood Web site), with a minimum threshold variant allele frequency (VAF) of 1% to 5% and 100–250X coverage. CR, CRi, and morphologic leukemia-free state (MLFS) are defined as per 2017 ELN AML recommendations.10 OS was calculated from the start of treatment to the date of death from any cause (cutoff date, 28 February 2019). Relapse-free survival was calculated from the date of remission (inclusive of CR, CRi, and MLFS) to the date of relapse or death from any cause. Both OS and relapse-free survival were censored for allogeneic stem cell transplantation or lost to follow-up. Kaplan-Meier survival curves were compared using log-rank statistics. All tests were 2 sided and considered significant where P < .05. Statistical analyses were performed using R statistical software version 3.6.0 (R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism version 8.
For details regarding experimental and molecular methods used in this research, refer to supplemental Methods.
Results
Defining prognostic subgroups among older patients with AML treated with venetoclax plus DNMTi or LDAC
A total of 81 patients treated with venetoclax in combination with DNMTi’s (azacitidine and decitabine; n = 58) or LDAC (n = 23) within previously reported clinical trials (NCT02287233 and NCT02203773) were included in the study cohort.4,5 These patients were treated at the MD Anderson Cancer Center (Houston) or the Alfred Hospital (Melbourne). The median age of this elderly cohort was 74 years (range, 62-87 years). The DNMTi + venetoclax study excluded prior DNMTi therapy. In contrast, the LDAC + venetoclax study included patients with prior DNMTi exposure. The target dose of venetoclax was also different in the 2 studies (DNMTi study, 400 mg/day; LDAC study, 600 mg/day).4,5 Clinical, cytogenetic, and molecular characteristics were balanced between the 2 groups (Table 1).
Overall CR/CRi was achieved in 69% in the DNMTi group and 52% in the LDAC group; comparable to outcomes reported in the parent clinical studies.4,5 Having confirmed this, we first sought to determine if the use of either DNMTi or LDAC influenced the outcome of newly diagnosed older patients with AML when combined with venetoclax. Despite a higher response rate with venetoclax plus DNMTi vs venetoclax plus LDAC, OS (Figure 1A; P = .52) and relapse-free survival (Figure 1B; P = .93) were similar, suggesting that the type of chemotherapy backbone was not a major influence on survival outcomes within this population. We therefore combined the treatment cohorts to maximize power to enable identification of clinical and molecular factors associated with treatment success and failure.
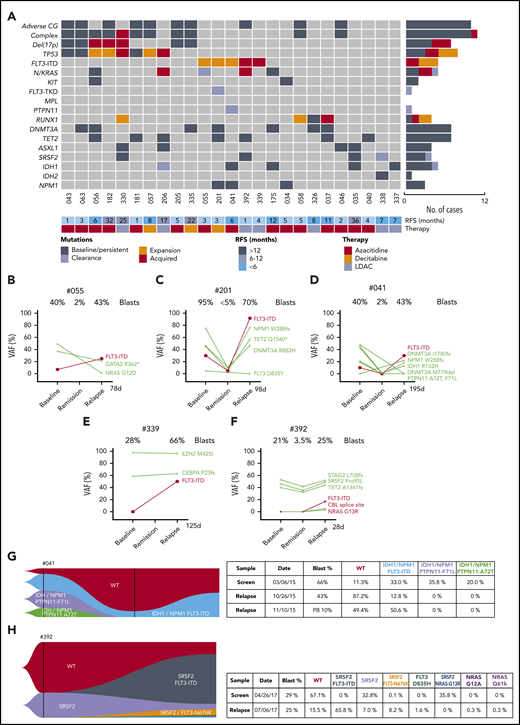
![Categorization of outcomes and genomic annotation of the study population. (A) Kaplan-Meier plots of OS for patients with AML treated with venetoclax in combination with either DNMTi’s or LDAC. n = 81, P = .52 (log-rank) for comparison between different combinations. (B) Kaplan-Meier plots for relapse-free survival for patients with AML achieving response (CR, n = 35; CRi, n = 17; and MLFS, n = 7) after treatment with either venetoclax in combination with DNMTi’s or LDAC. n = 59, P = .93 (log-rank) for comparison between different combinations. (C) Categorization of patients into those achieving durable remission (group A, n = 18), remission then relapse with adaptive resistance (group B, n = 25), primary refractory disease (group C, n = 20), or into an uncategorized group when the criteria were not met for any of the other categories (n = 18). The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, best response (CR, CRi, MLFS, resistant disease [RD], or nonevaluable [NE]), cytotoxic therapy received (AZA, azacitidine; DEC, decitabine; or LDAC), and prior exposure to DNMTi are shown for each case. Shown to the right of the plot are the number of cases achieving (dark gray) or not achieving (light gray) CR or CRi.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/11/10.1182_blood.2019003988/5/m_bloodbld2019003988f1.png?Expires=1770947967&Signature=dPaoQ2t3Ge24xnyFFByPPSKGc5Lx7fq5K47ogCu6nI33kK~2tQG8QzddncGmV7zjDHFZSObG96BOatbNQtv8SxpzzpOejwnC5cWLVnmQ-EIdp9LErfjB9NII3py3udTgLy7nrv7JYaVNyLP~ikXkHSiA6yJlrkiM2vLB~L1uXTiBhCi0N69aZ1691EFPjpEGF9y6oQzo6RchzGvxSgAe6Om8oJSgQdaGElS8YSKWKQ-84McKfgH3IM9CRyuyJH3QYmsZQgb9u8kCYkVIeyOd0tWIvZgyAsy5ha9TBD7UgPgz9AtuCHJ0VYTDX8s4pSk5iTu1ZPhdRBlMM6IRgfediw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Categorization of outcomes and genomic annotation of the study population. (A) Kaplan-Meier plots of OS for patients with AML treated with venetoclax in combination with either DNMTi’s or LDAC. n = 81, P = .52 (log-rank) for comparison between different combinations. (B) Kaplan-Meier plots for relapse-free survival for patients with AML achieving response (CR, n = 35; CRi, n = 17; and MLFS, n = 7) after treatment with either venetoclax in combination with DNMTi’s or LDAC. n = 59, P = .93 (log-rank) for comparison between different combinations. (C) Categorization of patients into those achieving durable remission (group A, n = 18), remission then relapse with adaptive resistance (group B, n = 25), primary refractory disease (group C, n = 20), or into an uncategorized group when the criteria were not met for any of the other categories (n = 18). The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, best response (CR, CRi, MLFS, resistant disease [RD], or nonevaluable [NE]), cytotoxic therapy received (AZA, azacitidine; DEC, decitabine; or LDAC), and prior exposure to DNMTi are shown for each case. Shown to the right of the plot are the number of cases achieving (dark gray) or not achieving (light gray) CR or CRi.
Categorization of outcomes and genomic annotation of the study population. (A) Kaplan-Meier plots of OS for patients with AML treated with venetoclax in combination with either DNMTi’s or LDAC. n = 81, P = .52 (log-rank) for comparison between different combinations. (B) Kaplan-Meier plots for relapse-free survival for patients with AML achieving response (CR, n = 35; CRi, n = 17; and MLFS, n = 7) after treatment with either venetoclax in combination with DNMTi’s or LDAC. n = 59, P = .93 (log-rank) for comparison between different combinations. (C) Categorization of patients into those achieving durable remission (group A, n = 18), remission then relapse with adaptive resistance (group B, n = 25), primary refractory disease (group C, n = 20), or into an uncategorized group when the criteria were not met for any of the other categories (n = 18). The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, best response (CR, CRi, MLFS, resistant disease [RD], or nonevaluable [NE]), cytotoxic therapy received (AZA, azacitidine; DEC, decitabine; or LDAC), and prior exposure to DNMTi are shown for each case. Shown to the right of the plot are the number of cases achieving (dark gray) or not achieving (light gray) CR or CRi.
Although increasing age is frequently associated with poorer prognosis in AML, survival among subjects ≥75 years was comparable to younger patients in this cohort, suggesting that advanced age alone was not a negative determinant of outcome with venetoclax-based therapies (supplemental Figure 1A). Adverse cytogenetic risk and prior DNMTi exposure, however, were associated with inferior survival (supplemental Figure 1B-C).
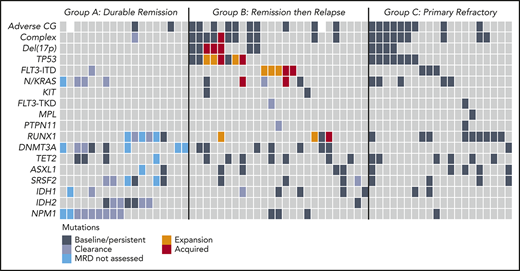
We used treatment response and duration of remission to define clinically relevant subgroups for investigation of molecular correlates of treatment sensitivity and resistance. Patients were categorized into those who experienced a sustained remission (>12 months) without relapse (group A; durable remission; n = 18), initial remission followed by relapse at any time indicating adaptive resistance (group B; response then relapse; n = 25), and patients refractory to primary therapy (group C; primary refractory; n = 20) (Figure 1C). There were 18 uncategorized patients who responded to therapy but received venetoclax-based therapy for <12 months for reasons other than relapse, including stem cell transplantation (n = 6), patient choice (n = 5), inadequate response (n = 3), and indeterminate causes of death or death in CR (n = 4). These patients were included in analyses of relapse-free survival and OS outcome (Figure 1A-B) but excluded from molecular associations with durable remission or adaptive resistance.
Molecular characteristics of patients with durable remission
In the whole population, venetoclax-based therapy induced CR/CRi in 64% of patients. The strongest molecular associations with response were noted in patients with NPM1, IDH1, IDH2, or DNMT3A mutations, with CR/CRi rates exceeding 80% (Figure 1C, right panel). To determine which mutations were associated with durable responses, we next focused on molecular characteristics linked to responses lasting for >12 months (cutoff date, 28 February 2019). A total of 18 patients with durable remission formed this subgroup (Figure 2A). NPM1 (9/18; 50%) and IDH2 (7/18; 39%) were among the most frequently mutated genes, with survival ongoing after 21 to 49 months follow-up (Figure 2A). NPM1mut was suppressed to undetectable levels in remission in 4 out of 4 cases assessed by highly sensitive quantitative reverse transcription polymerase chain reaction (RT-qPCR) that was sustained for at least 24 months (Figure 2B). In contrast, patients with IDH2mut had persistent high VAF levels in 3 out of 7 cases, whereas another 3 patients had molecular persistence at low VAF (0.03% to 0.11%) spanning 11 to 16 months; 1 patient had undetectable IDH2mut at 21 months (< 0.01%) (Figure 2C). The majority of patients with either NPM1 or IDH2 mutation resided in the durable remission cohort. Among patients with NPM1mut, only 4 resided in the relapsed and refractory subgroups (all had concurrent FLT3-ITD or KITmut). For patients with an IDH2mut, only 1 case has so far relapsed. Median OS for patients with either NPM1mut or IDH2mut was not reached, with 2-year OS of 71.8% and 79.5%, respectively (Figure 2D). In the durable remission group, DNMT3Amut was present in 44% of cases (8/18), and 6 out of 8 of these cases were among patients with concurrent NPM1 or IDH2mut. Therefore, mutant NPM1 and/or IDH2 was associated with a high response rate to treatment, durable remission, and prolonged OS.
![Characterization of AML cases with durable remission. (A) Genomic landscape of AML cases with durable remission. Molecular profile for 18 patients with ongoing remission for >12 months (date cutoff, 28 February 2019). The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, relapse-free survival (in months), and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine; or LDAC) are shown for each case. The bar graphs (right side of the plot) summarize the number of cases with persistent mutations or reduced/cleared mutations or where MRD was not assessed in remission. (B) Quantitative MRD profile of NPM1 mutation in 4 AML cases as assessed by RT-qPCR. MRD levels are expressed as percent relative to baseline transcript level. Positive detection is shown as solid-filled black circles, whereas negative detection is shown as open circles, indicating the assay sensitivity relative to amplification of the ABL gene. (C) Quantitative MRD profile of IDH2 mutation in 7 AML cases as assessed by Droplet Digital PCR (n = 5) and next-generation sequencing (NGS) panel (n = 2, both had high VAF). One additional case (#044) tested negative in remission by Sanger sequencing only and is not included in this figure. Morphologic relapse is indicated by an “R”. Undetectable IDH2mut level is indicated by “Neg”. (D) Kaplan-Meier plots of OS of patients with NPM1 or IDH2 mutant AML treated with venetoclax in combination with either DNMTi or LDAC compared with patients WT for both NPM1 and IDH2. Four patients had concurrent NPM1 and IDH2 mutations and were included twice. One patient was excluded due to unknown molecular status. (E) Kaplan-Meier plots of OS of patients with IDH1 mutant AML treated with venetoclax in combination with either DNMTi or LDAC compared with patients WT for IDH1. One patient was excluded due to unknown molecular status. (F) Volcano plot of differential gene expression associated with NPM1 mutation in responders: NPM1mut (n = 4) vs NPM1 WT (n = 6). No genes were significantly differentially expressed (DE) with FDR <5%. HOX genes are highlighted in green and apoptosis pathway genes in purple. Gene set enrichment analysis confirmed activation of the NPM1mut signature (using correlation adjusted mean rank gene set analysis [CAMERA], top 3 NPM1mut gene sets, FDR range from 5.17 × 10−21 to 2.51 × 10−27). (G) Volcano plot of differential gene expression associated with NPM1 mutation in TCGA dataset: NPM1mut (n = 35) vs NPM1 WT (n = 115). Gene sets are colored as in panel F, with differentially expressed genes in blue and red. Gene set enrichment analysis confirmed activation of the NPM1mut signature (using CAMERA, top 3 NPM1mut gene sets, FDR range 1.9 × 10−34 to 1.1 × 10−37).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/11/10.1182_blood.2019003988/5/m_bloodbld2019003988f2.png?Expires=1770947967&Signature=qQf1cb9f4ELCk1p1YRDxN368YTef5xxMO4gXwj0~BsEZ036qP6Dfh5n6aes7F3vHsWDEjBfh74O65XEP5tknmhq1TqEo9Mtrv7nwp0R9PygeacDt4Q9pNhiYMh0P7fCRQ9vrsj0VcxJFYg9NyI~eGdqGZaFV7cfLBxSME4vT5ofOmNhR149wFCLsl3vFTvVUgZ8ymlKJDepxNho1gj7rFdHScSbDiGqiTQFBMex-Y88yTotoWeAzcLOOtVJfXiP8QzX8nwwAbNJDWfUaczOXXil9hwhrc5ZDSJYR28FDf0WgvBVPICBmKs6odfqV7SPhOt6bmJ3z48KB~DBBDy9EiQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characterization of AML cases with durable remission. (A) Genomic landscape of AML cases with durable remission. Molecular profile for 18 patients with ongoing remission for >12 months (date cutoff, 28 February 2019). The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, relapse-free survival (in months), and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine; or LDAC) are shown for each case. The bar graphs (right side of the plot) summarize the number of cases with persistent mutations or reduced/cleared mutations or where MRD was not assessed in remission. (B) Quantitative MRD profile of NPM1 mutation in 4 AML cases as assessed by RT-qPCR. MRD levels are expressed as percent relative to baseline transcript level. Positive detection is shown as solid-filled black circles, whereas negative detection is shown as open circles, indicating the assay sensitivity relative to amplification of the ABL gene. (C) Quantitative MRD profile of IDH2 mutation in 7 AML cases as assessed by Droplet Digital PCR (n = 5) and next-generation sequencing (NGS) panel (n = 2, both had high VAF). One additional case (#044) tested negative in remission by Sanger sequencing only and is not included in this figure. Morphologic relapse is indicated by an “R”. Undetectable IDH2mut level is indicated by “Neg”. (D) Kaplan-Meier plots of OS of patients with NPM1 or IDH2 mutant AML treated with venetoclax in combination with either DNMTi or LDAC compared with patients WT for both NPM1 and IDH2. Four patients had concurrent NPM1 and IDH2 mutations and were included twice. One patient was excluded due to unknown molecular status. (E) Kaplan-Meier plots of OS of patients with IDH1 mutant AML treated with venetoclax in combination with either DNMTi or LDAC compared with patients WT for IDH1. One patient was excluded due to unknown molecular status. (F) Volcano plot of differential gene expression associated with NPM1 mutation in responders: NPM1mut (n = 4) vs NPM1 WT (n = 6). No genes were significantly differentially expressed (DE) with FDR <5%. HOX genes are highlighted in green and apoptosis pathway genes in purple. Gene set enrichment analysis confirmed activation of the NPM1mut signature (using correlation adjusted mean rank gene set analysis [CAMERA], top 3 NPM1mut gene sets, FDR range from 5.17 × 10−21 to 2.51 × 10−27). (G) Volcano plot of differential gene expression associated with NPM1 mutation in TCGA dataset: NPM1mut (n = 35) vs NPM1 WT (n = 115). Gene sets are colored as in panel F, with differentially expressed genes in blue and red. Gene set enrichment analysis confirmed activation of the NPM1mut signature (using CAMERA, top 3 NPM1mut gene sets, FDR range 1.9 × 10−34 to 1.1 × 10−37).
Characterization of AML cases with durable remission. (A) Genomic landscape of AML cases with durable remission. Molecular profile for 18 patients with ongoing remission for >12 months (date cutoff, 28 February 2019). The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, relapse-free survival (in months), and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine; or LDAC) are shown for each case. The bar graphs (right side of the plot) summarize the number of cases with persistent mutations or reduced/cleared mutations or where MRD was not assessed in remission. (B) Quantitative MRD profile of NPM1 mutation in 4 AML cases as assessed by RT-qPCR. MRD levels are expressed as percent relative to baseline transcript level. Positive detection is shown as solid-filled black circles, whereas negative detection is shown as open circles, indicating the assay sensitivity relative to amplification of the ABL gene. (C) Quantitative MRD profile of IDH2 mutation in 7 AML cases as assessed by Droplet Digital PCR (n = 5) and next-generation sequencing (NGS) panel (n = 2, both had high VAF). One additional case (#044) tested negative in remission by Sanger sequencing only and is not included in this figure. Morphologic relapse is indicated by an “R”. Undetectable IDH2mut level is indicated by “Neg”. (D) Kaplan-Meier plots of OS of patients with NPM1 or IDH2 mutant AML treated with venetoclax in combination with either DNMTi or LDAC compared with patients WT for both NPM1 and IDH2. Four patients had concurrent NPM1 and IDH2 mutations and were included twice. One patient was excluded due to unknown molecular status. (E) Kaplan-Meier plots of OS of patients with IDH1 mutant AML treated with venetoclax in combination with either DNMTi or LDAC compared with patients WT for IDH1. One patient was excluded due to unknown molecular status. (F) Volcano plot of differential gene expression associated with NPM1 mutation in responders: NPM1mut (n = 4) vs NPM1 WT (n = 6). No genes were significantly differentially expressed (DE) with FDR <5%. HOX genes are highlighted in green and apoptosis pathway genes in purple. Gene set enrichment analysis confirmed activation of the NPM1mut signature (using correlation adjusted mean rank gene set analysis [CAMERA], top 3 NPM1mut gene sets, FDR range from 5.17 × 10−21 to 2.51 × 10−27). (G) Volcano plot of differential gene expression associated with NPM1 mutation in TCGA dataset: NPM1mut (n = 35) vs NPM1 WT (n = 115). Gene sets are colored as in panel F, with differentially expressed genes in blue and red. Gene set enrichment analysis confirmed activation of the NPM1mut signature (using CAMERA, top 3 NPM1mut gene sets, FDR range 1.9 × 10−34 to 1.1 × 10−37).
The association between IDH1mut and prognosis was less clear. There were 2 IDH1mut cases in the durable remission subgroup, and both had a co-occurring NPM1mut (Figure 2A). Among the 7 IDH1mut cases occurring in patients with relapsing or primary refractory disease, 5 cases had a concurrent TP53, FLT3-ITD, or RAS mutation (Figure 1C). The median OS for patients with IDH1mut was not significantly different from patients with IDH1 wild-type (WT) AML (18.3 vs 12.7 months; P = .79) (Figure 2E).
To gain further insight into differentially expressed genes associated with long-term NPM1mut responders, we performed gene expression profiling on a subset of patients with NPM1mut (n = 4) compared with NPM1 WT (n = 6) (Figure 2F), focusing on differences in expression of genes encoding proteins that regulate apoptosis. The NPM1mut group was enriched in expression of HOX/MEIS1 gene family members, and gene set testing confirmed activation of an NPM1mut signature (CAMERA test; false discovery rate [FDR] range, 5.17 × 10−21 to 2.51 × 10−27) (supplemental Figure 2A). We searched for expression changes in key apoptosis pathway components in NPM1mut responders but detected no differences in our cohort using an FDR of <5%. Analysis of The Cancer Genome Atlas (TCGA) AML cohort (Figure 2G) confirmed activation of the NPM1mut signature and suggested modest differences in expression of apoptosis pathway components (eg, BCL2A1), but these changes were not identified within our cohort. Further analysis showed that the HOX gene expression signature was most closely linked to patients with an NPM1 mutation (supplemental Figure 2B). In contrast, there was no association between a HOX gene signature and patients with either an IDH or SRSF2 mutation.
Adaptive resistance to venetoclax combinations is associated with kinase activation
Twenty-five cases had adaptive resistance, representing 31% of the total cohort of 81 patients (Figure 3A). The median time to relapse was 6.4 months (95% confidence interval, 4.5-10.6 months); 5 patients relapsed after 12 months. To identify dynamic molecular changes indicative of adaptive resistance, the VAFs of individual mutations were compared at diagnosis, in remission, and at relapse to identify clones expanded at relapse. Two important findings emerged: progressive expansion of clones with activated kinases, particularly FLT3-ITD, and in other cases, selection of clones with likely biallelic perturbation of TP53.
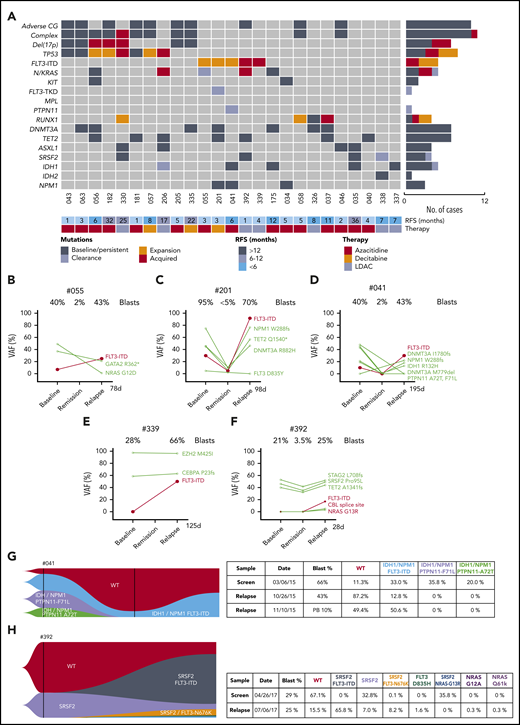
Characterization of AML cases with adaptive resistance. (A) Genomic landscape of AML cases with adaptive resistance. Molecular mutations in 25 patients with relapsed AML after an initial response. The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, relapse-free survival, and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine or LDAC, low-dose cytarabine) are shown for each case. The bar graphs (right side of the plot) summarize the number of cases with persistent, expanded, acquired, or reduced/cleared mutations at relapse. (B-F) Dynamic changes in clonal architecture from diagnosis to relapse in 5 cases with FLT3-ITD. The VAF for each mutation is shown, along with the bone marrow blast count at the corresponding time point. The time elapsed from remission to treatment failure is shown in days. (G-H) Single-cell analysis of clonal architecture at screening and relapse. Mission Bio Tapestri clonograms showing the relative mutation composition (%) of samples at indicated time points for cases #041 (G) and #392 (H). In case #041 (G), the NPM1 and IDH1 parental clone included 2 PTPN11mut subclones, which were extinguished, and a FLT3-ITD subclone, which expanded at relapse. In case #392 (H), the proportion of WT and SRSF2mut-only cells have decreased and been replaced by 4 new clones activating the FLT3 or NRAS kinase pathways.
Characterization of AML cases with adaptive resistance. (A) Genomic landscape of AML cases with adaptive resistance. Molecular mutations in 25 patients with relapsed AML after an initial response. The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, relapse-free survival, and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine or LDAC, low-dose cytarabine) are shown for each case. The bar graphs (right side of the plot) summarize the number of cases with persistent, expanded, acquired, or reduced/cleared mutations at relapse. (B-F) Dynamic changes in clonal architecture from diagnosis to relapse in 5 cases with FLT3-ITD. The VAF for each mutation is shown, along with the bone marrow blast count at the corresponding time point. The time elapsed from remission to treatment failure is shown in days. (G-H) Single-cell analysis of clonal architecture at screening and relapse. Mission Bio Tapestri clonograms showing the relative mutation composition (%) of samples at indicated time points for cases #041 (G) and #392 (H). In case #041 (G), the NPM1 and IDH1 parental clone included 2 PTPN11mut subclones, which were extinguished, and a FLT3-ITD subclone, which expanded at relapse. In case #392 (H), the proportion of WT and SRSF2mut-only cells have decreased and been replaced by 4 new clones activating the FLT3 or NRAS kinase pathways.
Serial analysis of diagnosis-remission-relapse samples identified an increased FLT3-ITD clonal burden in 5 patients at the time of disease progression (Figures 3B-F), with 2 cases displaying newly acquired FLT3-ITD (Figure 3E-F). The expansion or acquisition of FLT3-ITD in all 5 patients with adaptive resistance occurred within 1 to 6 months from initial remission (Figure 3A). One patient (#302) with FLT3-ITD (allele burden 28%) and concomitant NPM1 mutation achieved a durable remission (Figure 2A). Therefore, although initial responses to venetoclax-based combinations in patients with FLT3-ITD were comparable to the overall study population, relapse associated with clonal expansion of FLT3-ITD was a feature in this study cohort.
Single-cell sequencing technologies were employed to gain more detailed insight into the subclonal architecture and patterns of gene interactions not evident using bulk sequencing approaches. For patient #041, although bulk sequencing showed expansion of FLT3-ITD and clearance of cells bearing mutant PTPN11 (F71L and A72T) at relapse, single-cell sequencing additionally showed outgrowth of a triple-mutant FLT3-ITD/IDH1/NPM1 resistant subclone, whereas parallel PTPN11/IDH1/NPM1 subclones remained sensitive to treatment (Figure 3D,G). Therefore, single-cell sequencing was able to demonstrate the selective impact of FLT3-ITD in mediating resistance within clones harboring mutant NPM1 and IDH1. Single-cell sequencing was also informative in demonstrating the presence of actionable targets; namely, FLT3-ITD and IDH1 jointly present in 1 clone and only IDH1 in another.
A striking example of polyclonal resistance was observed in patient #392 using single-cell technology. In this case, after attaining remission with venetoclax-LDAC, early clinical progression occurred 1 month later, with bulk sequencing showing enrichment of 3 kinase mutations at relapse (FLT3-ITD, CBL, and NRAS) (Figure 3F). In addition to FLT3-ITD, single-cell analysis identified parallel expansion of 5 additional kinase bearing clones not identified on bulk sequencing (FLT3 N676K, NRAS G13R, FLT3 D835H, NRAS G12A, and NRAS Q61K) (Figure 3H). Interestingly, some of these kinases represented subclonal outgrowths of an SRSF2mut population, whereas other kinases expanded independently of SRSF2mut (Figure 3H). The SRSF2mut clone lacking a co-occurring kinase mutation contracted during therapy, in contrast to populations harboring an activated kinase.
In contrast to the polyclonal pattern of resistance, another pattern of venetoclax-based treatment failure involved clonal evolution of FLT3-ITD loss of heterozygosity (LOH) at relapse. In case #201, the FLT3-ITD allelic burden was ∼30% at diagnosis and suppressed to almost undetectable levels by venetoclax-decitabine therapy during remission (Figure 3C). Three months after first CR, the patient relapsed, with 70% bone marrow blasts and an increased FLT3-ITD burden of ∼90%, indicating likely loss of the WT FLT3 allele. Hitherto novel mechanisms of FLT3 activation were also identified at relapse. In case #339, FLT3-ITD was not present at diagnosis but emerged at relapse as detected by fragment analysis and capillary electrophoresis (Figure 3E). Interestingly, RNA sequencing (RNA-seq) identified an additional novel 10-base insertion in exon 23 of FLT3, resulting in C-terminal truncation of the protein. Cloning revealed this sequence resided in trans to the allele harboring FLT3-ITD in exon 14 (supplemental Figure 3A). Functional assessment in Ba/F3 cells showed that the truncated variant had a hypomorphic effect, leading to reduced protein stability and loss of signaling capacity (supplemental Figure 3B-C). Genetic studies in mouse models suggested that inactivation of the WT allele contributed to enhanced signaling via FLT3-ITD.11,12 Therefore, although the FLT3-ITD burden in this case was ∼50%, the remaining FLT3 allele was essentially hypoactive, rendering cells functionally homozygous for FLT3-ITD. In summary, our analysis of relapse samples from patients exposed to venetoclax-based therapeutic pressure revealed divergent mechanisms of resistance centered on the biological amplification of the FLT3-ITD signaling pathway.
FLT3-ITD causes dual resistance to both venetoclax and LDAC in vitro
To model the effect of FLT3-ITD on sensitivity to venetoclax and cytarabine, Ba/F3 cells were transduced to overexpress FLT3-ITD. When expressed in cells, FLT3-ITD resulted in cytokine-independent increases in BCL-XL and MCL1 protein expression not evident after transduced expression of an empty vector (supplemental Figure 4). Viability assays showed that FLT3-ITD rendered Ba/F3 cells resistant to venetoclax alone and also in combination with cytarabine (supplemental Figure 5). In contrast, FLT3-ITD–transduced cells were sensitive to FLT3 inhibitors, such as midostaurin or gilteritinib, with activity enhanced in combination with venetoclax. Addition of cytarabine as a “triplet” regimen did not appear to further enhance cytotoxicity over that observed for the venetoclax-FLT3 dual inhibitor combination (supplemental Figure 5). These studies highlight the potential for FLT3-ITD to drive expression of antiapoptotic proteins not targeted by venetoclax and the strong rationale for combination therapy with venetoclax and FLT3 inhibitors as a strategy to prevent FLT3-ITD–mediated resistance in AML.
Clones with biallelic TP53 defects enriched at relapse under selective venetoclax-chemotherapy pressure
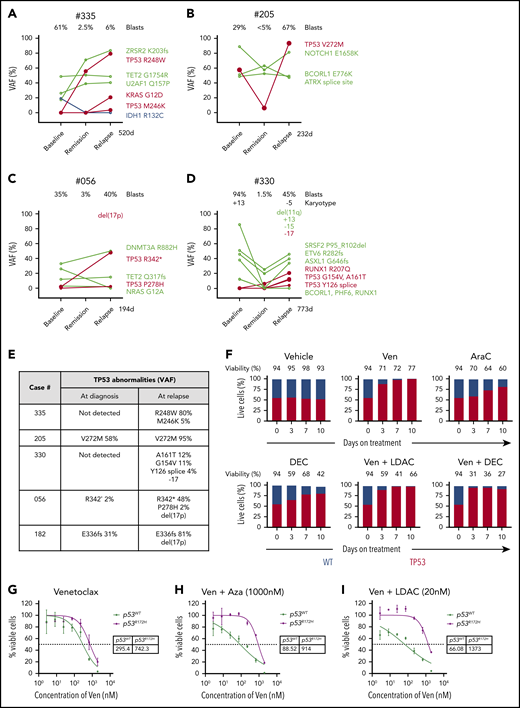
Mutant TP53 featured prominently in 8 out of 25 (32%) of the relapsing AML cohort, with 7 out of 8 TP53mut cases associated with complex karyotype (Figure 3A; supplemental Figure 6A). Strikingly, in 5 out of 5 cases with serial samples available, the size of the TP53mut clone increased under therapeutic pressure (Figure 4A-E), with 2 cases selecting for expansion of TP53 clones not detected at diagnosis (#335 and #330). In 3 cases, expansion of the TP53 mutation was accompanied by structural loss of the chromosome 17p locus, suggesting evolution of biallelic TP53 perturbations at relapse (Figure 4E). In cases #335 (TP53 R248W) and #205 (TP53 V272M), the TP53 VAF rose to 80% to 95%, indicating selection of cells with loss of the WT allele at relapse (Figure 4A-B,E). For case #335, TP53 R248W at relapse (VAF 80%) was accompanied by a contemporaneous rise in KRAS G12D (VAF 21%), suggesting the possibility of a cooperative interaction between these 2 driver mutations and venetoclax resistance (Figure 4A). However, for case #056, the rise in TP53 R342* at relapse was accompanied by the diminution of NRAS G12A (Figure 4C), suggesting the effect of activating RASmut as a contributor to venetoclax resistance was not consistent. Similar to activation of kinase pathways, polyclonal selection of clones with biallelic TP53 abnormalities were observed at relapse. For example, in cases #335, #330, and #056, additional TP53 variants emerged, suggesting the likelihood of multiple clones, with some likely to contain biallelic TP53 abnormalities (Figure 4E).
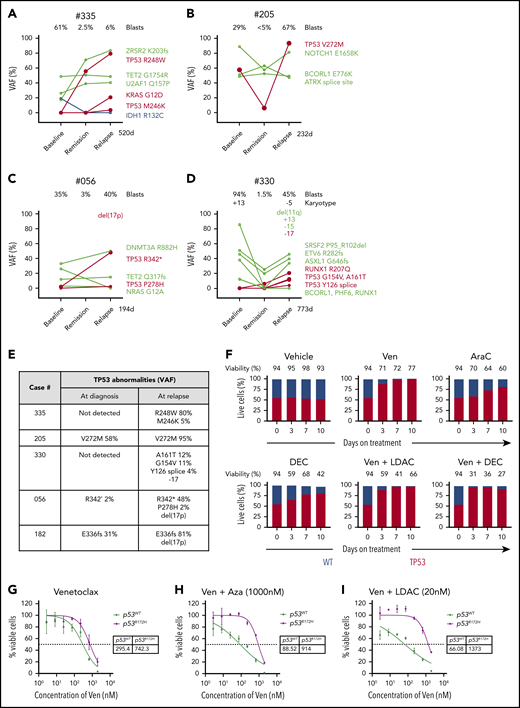
Impact of TP53 defects on venetoclax combinations. (A-D) Dynamic changes in clonal architecture from diagnosis to relapse in 4 illustrative cases with TP53 mutations. The VAF of each mutation is shown, along with the bone marrow blast count at the corresponding time point. The time elapsed from remission to treatment failure is shown in days. (E) Interval changes in chromosome 17 and TP53 at diagnosis and relapse. Patterns of clonal evolution of biallelic TP53 abnormalities in 5 AML cases at relapse. Changes in TP53 VAF % were quantitated by targeted NGS. Changes in chromosome 17 were assessed using standard karyotypic techniques. (F) Competitive growth assay comparing the survival of TP53 WT (gray) and TP53 CRISPR/Cas9–deleted (red) in MV4;11 cells during exposure to vehicle, venetoclax 100 nM, cytarabine (ara-C) 500 nM, decitabine 1 µM, venetoclax 100 nM plus low-dose cytarabine (LDAC) 100 nM, or venetoclax 100 nM plus decitabine (DEC) 1 µM in culture over a period of 10 days. The proportion of each cell genotype was indicated by a fluorescent reporter and enumerated by flow cytometry. The total cell viability (%) is shown above each bar. (G-I) Viability of RN2 cells (TP53 WT, p53WT) or p53R172H/Δ murine AML cell lines after exposure to venetoclax (G), venetoclax plus azacitidine (1 μM) (H), or venetoclax plus cytarabine (20 nM) (I) for 48 hours. The tables indicate the 50% inhibitory concentration values (nM). Errors represent the standard deviation of 3 technical replicates.
Impact of TP53 defects on venetoclax combinations. (A-D) Dynamic changes in clonal architecture from diagnosis to relapse in 4 illustrative cases with TP53 mutations. The VAF of each mutation is shown, along with the bone marrow blast count at the corresponding time point. The time elapsed from remission to treatment failure is shown in days. (E) Interval changes in chromosome 17 and TP53 at diagnosis and relapse. Patterns of clonal evolution of biallelic TP53 abnormalities in 5 AML cases at relapse. Changes in TP53 VAF % were quantitated by targeted NGS. Changes in chromosome 17 were assessed using standard karyotypic techniques. (F) Competitive growth assay comparing the survival of TP53 WT (gray) and TP53 CRISPR/Cas9–deleted (red) in MV4;11 cells during exposure to vehicle, venetoclax 100 nM, cytarabine (ara-C) 500 nM, decitabine 1 µM, venetoclax 100 nM plus low-dose cytarabine (LDAC) 100 nM, or venetoclax 100 nM plus decitabine (DEC) 1 µM in culture over a period of 10 days. The proportion of each cell genotype was indicated by a fluorescent reporter and enumerated by flow cytometry. The total cell viability (%) is shown above each bar. (G-I) Viability of RN2 cells (TP53 WT, p53WT) or p53R172H/Δ murine AML cell lines after exposure to venetoclax (G), venetoclax plus azacitidine (1 μM) (H), or venetoclax plus cytarabine (20 nM) (I) for 48 hours. The tables indicate the 50% inhibitory concentration values (nM). Errors represent the standard deviation of 3 technical replicates.
TP53 loss promotes resistance to both venetoclax and chemotherapy
To model the effect of TP53 mutation on sensitivity to venetoclax and DNMTi/cytarabine, clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 was used to disrupt TP53 in MV4;11 cells. Successful disruption of the TP53 locus was confirmed by sequencing and functional resistance to an MDM2 inhibitor. Distinctive fluorescent labels allowed flow cytometric quantitation of the proportion of CRISPR/Cas9 TP53 deleted (BFP) and WT cells (GFP) in culture over a period of 10 days (Figures 4F). Under selective therapeutic pressure, competitive outgrowth of TP53 defective vs WT cells was evident during exposure to venetoclax, cytarabine, or decitabine. Furthermore, resistance of TP53-defective cells was not abrogated by combination of venetoclax with either cytarabine or DNMT3i.
The role of TP53 biallelic mutation in resistance to combination treatment was confirmed using a murine AML model expressing a p53 R172H gain-of-function mutant.13,14 While there was only a marginal effect on sensitivity to venetoclax alone (Figure 4G), resistance to the combination of venetoclax with either azacitidine or cytarabine was striking (Figures 4H-I). These results confirm our finding that TP53 dysfunction or loss blunts the effectiveness of venetoclax-based combination therapy in AML.
Molecular patterns of primary resistance to venetoclax chemotherapy
Analysis of patients with primary refractory AML revealed 3 patterns of resistance: TP53mut, RUNX1mut, and activating kinase mutations. One-third (7/20) of the primary refractory cases harbored TP53mut (Figure 5A). In the majority of these cases (4/7), del(17p) was also present, indicating a biallelic TP53 abnormality was likely present at diagnosis (Figure 5A). Of the 3 primary refractory cases without del(17p), 2 had <1 TP53 abnormality, suggesting the potential for biallelic TP53 perturbation. Case #176 harbored both TP53 K120R (VAF 22%) and C238Y (12%), whereas case #053 had TP53 C242* (VAF 44%) and V272M (VAF 51%) (see Figure 5B). The third refractory AML case (#346) displayed a TP53 mutation (R175H) at high tumoral burden (VAF 81%), suggesting TP53 LOH. Concordant with our observations that biallelic TP53 abnormalities were associated with venetoclax-based resistance, the magnitude of aberrant TP53 at baseline was highest for patients with primary refractory disease, followed by patients with early relapsing disease, whereas TP53mut was mostly undetectable at diagnosis in patients with late relapse (Figure 5C).
![Characterization of AML cases with primary resistance. (A) Genomic landscape of AML cases with primary resistance. Molecular profile of 20 AMLs that were refractory to venetoclax-based combinations. The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine or LDAC, low-dose cytarabine) are shown for each case. Mutations detected by RNA-seq are represented by a dark gray box. For case #353, the KIT-ITD increased in frequency from 0.9% to 27% VAF from screening to after cycle 1 assessment. (B) TP53 abnormalities at baseline in refractory AML cases. TP53 VAFs % were quantitated by targeted NGS. (C) Baseline VAF of TP53 mutations according to patients with late relapse (>12 months), early relapse (<12 months), or primary refractory to treatment. AMLs with baseline and acquired del(17p) are indicated by orange and red, respectively. Each patient in a group is represented by a symbol. Some AML cases had >1 TP53 mutation: late relapse (#182 [inverted triangle], #335 [open circle] and #330 [upright triangle]); early relapse (#056 [square]); and refractory disease (#053 [hexagon], #176 [black circle] and #212 [upright triangle]). (D) Single-cell analysis of clonal architecture at screening and after treatment. Mission Bio Tapestri clonogram of case #064 showing the relative mutation composition (%) of samples at patient screening or at the time of refractory disease. In this case, within the EZH2mut clone, 5 parallel RASmut subclones are shown to emerge, with concurrent suppression of EZH2mut-only cells.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/11/10.1182_blood.2019003988/5/m_bloodbld2019003988f5.png?Expires=1770947967&Signature=tdWbV9ziRR-tCYTcnoQ4wfbvNCGeLpEg7CA26ICPJioBRk3LiFhVgDMzUEPQnZOCPUmBojw1uLOUaP36WS9NmohZ-7cy7Sh4C1lfJuSwXjljYwf0Bcrhcd3nkHMbN69~FCory~spZtDR0x1vy0kq8aIGdSZbswqtboPxDobdtZNSh9eEDpXd8lB5WoNjpandN8I0ZfCJsKtuZZt4zK4zDl7g1huR33RQ1b~SyJzxY4-0DDgxYYHaimYQYfl7kkTYu7uTp0hekz4CiyDDpyqExWcxI36eoN3MrSB-9Wuab7RrlVowi5td5o5rlrtxP0O0Yq35zPFeY6HmaahvrVzFDA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Characterization of AML cases with primary resistance. (A) Genomic landscape of AML cases with primary resistance. Molecular profile of 20 AMLs that were refractory to venetoclax-based combinations. The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine or LDAC, low-dose cytarabine) are shown for each case. Mutations detected by RNA-seq are represented by a dark gray box. For case #353, the KIT-ITD increased in frequency from 0.9% to 27% VAF from screening to after cycle 1 assessment. (B) TP53 abnormalities at baseline in refractory AML cases. TP53 VAFs % were quantitated by targeted NGS. (C) Baseline VAF of TP53 mutations according to patients with late relapse (>12 months), early relapse (<12 months), or primary refractory to treatment. AMLs with baseline and acquired del(17p) are indicated by orange and red, respectively. Each patient in a group is represented by a symbol. Some AML cases had >1 TP53 mutation: late relapse (#182 [inverted triangle], #335 [open circle] and #330 [upright triangle]); early relapse (#056 [square]); and refractory disease (#053 [hexagon], #176 [black circle] and #212 [upright triangle]). (D) Single-cell analysis of clonal architecture at screening and after treatment. Mission Bio Tapestri clonogram of case #064 showing the relative mutation composition (%) of samples at patient screening or at the time of refractory disease. In this case, within the EZH2mut clone, 5 parallel RASmut subclones are shown to emerge, with concurrent suppression of EZH2mut-only cells.
Characterization of AML cases with primary resistance. (A) Genomic landscape of AML cases with primary resistance. Molecular profile of 20 AMLs that were refractory to venetoclax-based combinations. The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine or LDAC, low-dose cytarabine) are shown for each case. Mutations detected by RNA-seq are represented by a dark gray box. For case #353, the KIT-ITD increased in frequency from 0.9% to 27% VAF from screening to after cycle 1 assessment. (B) TP53 abnormalities at baseline in refractory AML cases. TP53 VAFs % were quantitated by targeted NGS. (C) Baseline VAF of TP53 mutations according to patients with late relapse (>12 months), early relapse (<12 months), or primary refractory to treatment. AMLs with baseline and acquired del(17p) are indicated by orange and red, respectively. Each patient in a group is represented by a symbol. Some AML cases had >1 TP53 mutation: late relapse (#182 [inverted triangle], #335 [open circle] and #330 [upright triangle]); early relapse (#056 [square]); and refractory disease (#053 [hexagon], #176 [black circle] and #212 [upright triangle]). (D) Single-cell analysis of clonal architecture at screening and after treatment. Mission Bio Tapestri clonogram of case #064 showing the relative mutation composition (%) of samples at patient screening or at the time of refractory disease. In this case, within the EZH2mut clone, 5 parallel RASmut subclones are shown to emerge, with concurrent suppression of EZH2mut-only cells.
Another treatment-refractory subgroup largely mutually exclusive to the TP53mut subgroup comprised RUNX1 mutations in 40% of primary refractory cases (Figure 5A). Mutations in RUNX1 were located predominantly in the RUNT homology and transactivation domains (supplemental Figure 6B). Of the 8 RUNX1mut cases, 4 had ≥1 co-occurring activated signaling pathway mutation (FLT3-ITD [n = 2], FLT3-TKD, N/KRAS, MPL, and PTPN11) (Figure 5A). RUNX1mut was also expanded or acquired at relapse in 3 patients (Figure 3A). The significance of RUNX1mut as a driver of treatment resistance remains uncertain, as RUNX1mut cases were present among one-third of the 18 patients with long remission; notably, each of these cases harbored either a co-occurring IDH2 and/or SRSF2 mutation (Figure 2A). The role of RUNX1mut in mediating resistance to venetoclax will require further study in a larger patient cohort. There were 4 primary refractory cases outside the TP53 and RUNX1 mutant groups, and each case harbored an activating kinase mutation (FLT3-ITD, N/KRAS, CBL, or KIT). In total, among the 20 refractory cases, a signaling pathway mutation was present in 12 (60%), in contrast to 27% in the durable-remission group.
While we consider patients to have morphologically refractory disease based on assessment of total blast counts alone, interval molecular profiling before and after only 1 cycle of therapy revealed a surprising degree of intratumoral clonal heterogeneity not evident from examination of bulk blast levels alone. For example, among the primary refractory group, 1 case (#209) was double positive for NPM1mut and FLT3-ITD (Figure 5A). After venetoclax-azacitidine therapy, despite no evidence of a blast response, the NPM1mut level was reduced from 100% to 7.3% during the first cycle (as assessed by RT-qPCR), suggesting a discordant response to therapy. In another refractory AML case, single-cell analysis of case #064 revealed polyclonal expansion of 5 independent RAS clones (3 NRAS and 2 KRAS), with each clone associated with a co-occurring EZH2 mutation (Figure 5D). Although the RAS-containing clones expanded, an ancestral EZH2mut clone lacking subclonal RAS mutations was suppressed by therapy. In case #353, serial RNA-seq during the first cycle revealed expansion (VAF 0.9% to 27%) of a new KIT-ITD–bearing clone, with duplication of a 17-amino-acid segment (D572-R588). These findings highlight the potential for rapid, polyclonal, and divergent changes in clonal architecture, even among patients with “no morphologic response” to treatment. These findings reinforce the value of serial molecular studies to identify patterns of drug sensitivity and resistance at the subclonal level, even among patients with morphologically “refractory disease.”
Discussion
New drugs are entering the clinic for AML at a rapid rate. These include therapies targeting FLT3mut (midostaurin and gilteritinib), IDHmut (ivosidenib and enasidenib), hedgehog (glasdegib), or BCL-2 (venetoclax). High response rates achieved with venetoclax combinations have led to widespread use of these regimens as frontline therapy in older patients unfit for intensive chemotherapy.4,5,15 With availability of an increased therapeutic armamentarium for patients with AML, insight into patterns of response and resistance will be essential to guide optimal therapy and rationally design future clinical research studies.
Our study has clearly identified 2 major mechanisms of resistance to venetoclax-based combination therapy: activated kinase signaling and biallelic TP53 perturbation. There was surprising diversity in the range of kinase activation pathways affected. In addition to selection of FLT3-ITD, we found that kinase activation could be polyclonal with coactivation of related signaling pathways, including FLT3-TKD, FLT3 N676, RASmut, or CBLmut. Selection of rare and novel kinase variants were also identified, including KIT-ITD and a FLT3 C-terminal hypomorphic deletion acquired in trans to FLT3-ITD. The complexity of polyclonal resistance will likely pose clinical challenges when considering salvage treatment options. Emergence of FLT3-TKD would predict resistance to type 2 FLT3 inhibitors (sorafenib or quizartinib),16 FLT3 N676K midostaurin resistance,17 and RAS mutation resistance to gilteritinib.6 In another example, relapse was associated with emergence of IDH1mut and FLT3-ITD in the same clone, indicating the need for either an IDH1 or FLT3 inhibitor (or both) to be considered.
The association between TP53 perturbation and resistance to venetoclax combined with DNMTi or cytarabine was also strong. Although the presence of monoallelic del(17p) or TP53mut did not preclude remission being initially attained, the presence of biallelic TP53 defects at diagnosis in patients with refractory disease and evolution of clones with biallelic TP53 abnormalities were common in patients at relapse. Using CRISPR/Cas9 to disrupt TP53 function in AML cell lines, we showed that TP53 loss mediated therapeutic resistance to venetoclax, DNMTi, and cytarabine alone as well as in combination. Our findings are supported by recent preclinical studies using a genome-wide CRISPR/Cas9 screen in leukemic cell lines identifying venetoclax resistance to be associated with defects in apoptotic pathway members (TP53, BAX, and PMAIP1).18,19 Our studies therefore indicate the importance of surveying genetic TP53 integrity both at diagnosis and in remission in order to identify patients likely to develop resistance to venetoclax-based therapy. Our results also indicate the future need to develop more effective therapeutic approaches to mitigate against TP53 mutant AML, both at the time of initial therapy and at treatment salvage.
Our research findings also identified genetic aberrations associated with favorable long-term prognosis with venetoclax-based therapy. For NPM1mut AML, a high rate of durable remission and survival after treatment was observed. MRD negativity using a highly sensitive RT-qPCR method was common and sustained for over 24 months (Figure 2B). The 2-year OS exceeding 70% for elderly patients with NPM1mut AML treated with venetoclax plus DNMTi or LDAC compares favorably with azacitidine (2-year OS <25%) or intensive chemotherapy (2-year OS < 40%), suggesting that venetoclax may be the preferred therapeutic option for this particular AML subgroup.20,21 Expression analyses did not reveal an obvious link between mutations in NPM1 and differential expression of apoptosis pathway genes. It has previously been reported that NPM1 mutations in AML were associated with a HOX gene expression signature, and expression of HOX genes has also been linked to BCL-2 inhibitor responsiveness.22 In our study, while HOX expression was clearly linked to mutations in NPM1, there was no clear link with mutations in IDH, which were also associated with high response rate to venetoclax. Furthermore, some patients that were refractory also showed activation of HOX genes. Consequently, we conclude that HOX overexpression is neither sufficient nor necessary for AML to be exquisitely sensitive to this drug. Further investigations will be required to identify the mechanistic link between NPM1mut and heightened susceptibility to venetoclax. In IDHmut AML, we found that survival was particularly favorable for IDH2mut. Despite the high clinical response rate of IDHmut AML to venetoclax in combination with DNMTi or LDAC, molecular persistence of IDHmut was common, suggesting that other approaches, such as IDH inhibitor maintenance therapy, may be required to extinguish residual IDHmut clones.
In summary, venetoclax in combination with conventional low-intensity approaches is a promising initial therapy for older patients with AML. Our studies highlight clinically relevant molecular correlates of outcome that will impact future treatment strategies and combination approaches. Baseline molecular characterization may allow patients to be risk stratified into a favorable risk NPM1mut subgroup where molecular MRD monitoring and even consideration of treatment cessation could be employed within a future clinical trial. Patients with IDHmut could be considered for postremission IDH inhibitor maintenance-based approaches aimed at eradicating residual molecular disease, and patients with FLT3-ITD mutations could benefit from the addition of targeted FLT3 inhibitors to prevent relapse. Novel clinical trials incorporating new agents targeting TP53mut/del(17p) drug-resistant clones remains an area of high unmet need. Our work also highlights the clinical value of single-cell technologies for surveying intraclonal patterns of response to therapy and the potential advantages such techniques might have for treatment evaluation compared with bulk sequencing approaches. In conclusion, our studies highlight an exciting and dynamic new era in the management of older patients with AML. New drugs and combinations, risk stratification, precision-based monitoring, and molecularly guided risk-adaptive therapy will likely be the new norm for older patients, who previously had few effective treatment options and where molecular characterization was generally considered futile.
DNA variant calls are available from the Illumina myeloid panel. RNA-seq data will be made available through the European Genome-phenome Archive (EGAS00001003820). The use of the sequencing data are subject to a data transfer agreement and is restricted to ethically approved research into blood cell malignancies and cannot be used to assess germline variants.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The results include analysis based on data generated by TCGA Research Network (http://cancergenome.nih.gov/).
This work was supported in part by grants from the National Health and Medical Research Council of Australia (1162809, 126843, 1113133, and 1113577), Leukemia and Lymphoma Society Specialized Centre of Research (SCOR- CI Strasser), Independent Research Institutes (Infrastructure Support Scheme grant 9000220), the Cancer Council Victoria (grant-in-aid 1124178) (I.J.M.), a Victorian State Government Operational Infrastructure Support grant, and fellowship support from the Victorian Cancer Agency (I.J.M.), the National Health and Medical Research Council of Australia (D.C.S.H., A.W.R.), the Leukemia Lymphoma Society (R.T.), the Felton Bequest (I.J.M.), the and Medical Research Future Fund (1141460) (A.H.W.). Scholarship support was provided by a Melbourne University Research Scholarship (A.Q.). This research is additionally supported in part by National Cancer Institute, National Institutes of Health cancer center support grant P30 CA016672 and the V Foundation Lloyd Family Clinical Scholar Award (C.D.D.).
Authorship
Contribution: A.H.W., C.D.D., I.S.T., A.W.R., I.J.M., and M.K. designed research, performed research, analyzed data, and wrote the paper; and A.Q., S.M., S.L., F.C.B., R.T., G.P., A.I., J.M.S., C.G., S.A.F., Q.Z., H.M., K.P.P., C.C.C., P.B., C.F., A.I., H.K., D.C.S.H., S.M.K., and Z.X. performed research, analyzed data, and contributed vital new reagents or analytical tools.
Conflict-of-interest disclosure: I.J.M., D.C.S.H., and A.W.R. are employees and A.W. a former employee of the Walter and Eliza Hall Institute, which receives milestone and royalty payments related to venetoclax. I.J.M., D.C.S.H., A.W.R., and A.H.W. received payments from the Walter and Eliza Hall Institute related to venetoclax. A.H.W. is a medical advisor and receives research funding and honoraria from Abbvie. C.D.D. has received research funding from AbbVie/Genentech, Agios, Celgene, and Daiichi Sankyo and served in a consultancy/advisory role for AbbVie, Agios, Celgene, Daiichi-Sankyo, Jazz and Notable Labs.
Correspondence: Andrew H. Wei, Department of Haematology, The Alfred Hospital, Australian Centre for Blood Diseases, Monash University, Melbourne, VIC 3004, Australia; e-mail: andrew.wei@monash.edu.

![Categorization of outcomes and genomic annotation of the study population. (A) Kaplan-Meier plots of OS for patients with AML treated with venetoclax in combination with either DNMTi’s or LDAC. n = 81, P = .52 (log-rank) for comparison between different combinations. (B) Kaplan-Meier plots for relapse-free survival for patients with AML achieving response (CR, n = 35; CRi, n = 17; and MLFS, n = 7) after treatment with either venetoclax in combination with DNMTi’s or LDAC. n = 59, P = .93 (log-rank) for comparison between different combinations. (C) Categorization of patients into those achieving durable remission (group A, n = 18), remission then relapse with adaptive resistance (group B, n = 25), primary refractory disease (group C, n = 20), or into an uncategorized group when the criteria were not met for any of the other categories (n = 18). The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, best response (CR, CRi, MLFS, resistant disease [RD], or nonevaluable [NE]), cytotoxic therapy received (AZA, azacitidine; DEC, decitabine; or LDAC), and prior exposure to DNMTi are shown for each case. Shown to the right of the plot are the number of cases achieving (dark gray) or not achieving (light gray) CR or CRi.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/11/10.1182_blood.2019003988/5/m_bloodbld2019003988f1.png?Expires=1770947968&Signature=rgp-SWY4QAMh99HtwBDorxVXbGXRazWPfccrDQ5GOvyQlaOFAoZIlF0Ben~SA-y~uNsOtaJu-yCpV7rUKUbh6erk77oArlafahFIusqnhGRgQraFblPZnknOUYdgrtB2Uq2tn72Ct36~ngv-apOP8PcTo-YpYZvqm3BfUlbj525EPzcz7RgIDpfaXQqjJyznmB0W8uy8HGB~FrVzeERUWd01FHvz0ZApbOf68RrUqvnWAC2C-lyc-9osmUGCejO1aHA7OXgrkB~kJfOEnliUJeOm5GHmV9Y1C2wvQQrRfN5yRP6BKbghlV5-mym9gv5RNZWkyBVvRiD7NuAo7mCeTw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Characterization of AML cases with durable remission. (A) Genomic landscape of AML cases with durable remission. Molecular profile for 18 patients with ongoing remission for >12 months (date cutoff, 28 February 2019). The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, relapse-free survival (in months), and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine; or LDAC) are shown for each case. The bar graphs (right side of the plot) summarize the number of cases with persistent mutations or reduced/cleared mutations or where MRD was not assessed in remission. (B) Quantitative MRD profile of NPM1 mutation in 4 AML cases as assessed by RT-qPCR. MRD levels are expressed as percent relative to baseline transcript level. Positive detection is shown as solid-filled black circles, whereas negative detection is shown as open circles, indicating the assay sensitivity relative to amplification of the ABL gene. (C) Quantitative MRD profile of IDH2 mutation in 7 AML cases as assessed by Droplet Digital PCR (n = 5) and next-generation sequencing (NGS) panel (n = 2, both had high VAF). One additional case (#044) tested negative in remission by Sanger sequencing only and is not included in this figure. Morphologic relapse is indicated by an “R”. Undetectable IDH2mut level is indicated by “Neg”. (D) Kaplan-Meier plots of OS of patients with NPM1 or IDH2 mutant AML treated with venetoclax in combination with either DNMTi or LDAC compared with patients WT for both NPM1 and IDH2. Four patients had concurrent NPM1 and IDH2 mutations and were included twice. One patient was excluded due to unknown molecular status. (E) Kaplan-Meier plots of OS of patients with IDH1 mutant AML treated with venetoclax in combination with either DNMTi or LDAC compared with patients WT for IDH1. One patient was excluded due to unknown molecular status. (F) Volcano plot of differential gene expression associated with NPM1 mutation in responders: NPM1mut (n = 4) vs NPM1 WT (n = 6). No genes were significantly differentially expressed (DE) with FDR <5%. HOX genes are highlighted in green and apoptosis pathway genes in purple. Gene set enrichment analysis confirmed activation of the NPM1mut signature (using correlation adjusted mean rank gene set analysis [CAMERA], top 3 NPM1mut gene sets, FDR range from 5.17 × 10−21 to 2.51 × 10−27). (G) Volcano plot of differential gene expression associated with NPM1 mutation in TCGA dataset: NPM1mut (n = 35) vs NPM1 WT (n = 115). Gene sets are colored as in panel F, with differentially expressed genes in blue and red. Gene set enrichment analysis confirmed activation of the NPM1mut signature (using CAMERA, top 3 NPM1mut gene sets, FDR range 1.9 × 10−34 to 1.1 × 10−37).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/11/10.1182_blood.2019003988/5/m_bloodbld2019003988f2.png?Expires=1770947968&Signature=BrkP8MrWWpsdK9H3t4RlEhptCEkJk9K63jKJeTv0gk2nKA-XGSx-LXnj4NOa6-2BcRLRoAyPbz0vu97GHzUjJIhU2~YyJiRH1eg-~cNReUVeVpKVVvpe9wudWfN8CX0DIiJJu2As47zKPhC61xgM90AFkrHZ-oLy-jx4yctscUOzClKUdW8bdFO2LAHHe89NqNOOrJHvE3HmnXZJInaTfMCdIoZeaWrGT5j~T3FDNS-uUesPRroykESoKdERZFapo48idjeHbOpxTF2ZddfbypNZdfF1m7JnI-Zh9uK6ppbFpBrm0F4v7wcDnk8eOHfuRY0r1sEI-1GEdaaExD1NYw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Characterization of AML cases with primary resistance. (A) Genomic landscape of AML cases with primary resistance. Molecular profile of 20 AMLs that were refractory to venetoclax-based combinations. The presence of adverse cytogenetic risk, complex karyotype, del(17p), indicated mutations, study ID number, and cytotoxic therapy received (AZA, azacitidine; DEC, decitabine or LDAC, low-dose cytarabine) are shown for each case. Mutations detected by RNA-seq are represented by a dark gray box. For case #353, the KIT-ITD increased in frequency from 0.9% to 27% VAF from screening to after cycle 1 assessment. (B) TP53 abnormalities at baseline in refractory AML cases. TP53 VAFs % were quantitated by targeted NGS. (C) Baseline VAF of TP53 mutations according to patients with late relapse (>12 months), early relapse (<12 months), or primary refractory to treatment. AMLs with baseline and acquired del(17p) are indicated by orange and red, respectively. Each patient in a group is represented by a symbol. Some AML cases had >1 TP53 mutation: late relapse (#182 [inverted triangle], #335 [open circle] and #330 [upright triangle]); early relapse (#056 [square]); and refractory disease (#053 [hexagon], #176 [black circle] and #212 [upright triangle]). (D) Single-cell analysis of clonal architecture at screening and after treatment. Mission Bio Tapestri clonogram of case #064 showing the relative mutation composition (%) of samples at patient screening or at the time of refractory disease. In this case, within the EZH2mut clone, 5 parallel RASmut subclones are shown to emerge, with concurrent suppression of EZH2mut-only cells.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/11/10.1182_blood.2019003988/5/m_bloodbld2019003988f5.png?Expires=1770947968&Signature=Z6gm1TwHbo7p8nmFHRwaw3rLLZnj3d8soaaNC~m2qvI0xAT9jmdM914Cw06UK9fQ5QWQPHbrpo94yCYgsEzwSJBtGLMRkgw7R1QqFRYzlZpMN4cHeC9psDkWC2tcL6Eohnryw3MsHF5DQR5V35g8qka8mrIDfXHffd87WrtMv2um4o2Llj1-arDR4oV0bjieTzXimAP9ZLjIatYWPpfVsyjXnJjFQ7E37D04Rh5D~uzuL30JpedNaYzVQU-0KMWXaLXej0QqyXdQWh624rFYeJvjZKvA7BUVscOLnfftST2UE2RYJd821JGU2R8cm6qAI043r27WrEsivVaQnivAaw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
