

TO THE EDITOR:
Ticagrelor, a reversible inhibitor of the adenosine 5′-diphosphate (ADP) receptor P2Y12, is recommended as a first-line P2Y12 receptor antagonist after coronary interventions or acute coronary syndrome.1,2 In contrast to the irreversible inhibitors aspirin, clopidogrel, and prasugrel, ticagrelor binds to the ADP receptor reversibly, and the drug is present at high concentrations in plasma (227-770 ng/mL).3
Heparin-induced thrombocytopenia (HIT) is a serious adverse reaction that is caused by platelet-activating anti–platelet factor 4 (PF4)/heparin antibodies and leads to an increased risk for thrombosis4 . The most widely used system for clinical diagnosis is the 4Ts score (0-8 points).5 The diagnosis is confirmed by laboratory tests. Antigen tests for anti-PF4/heparin antibodies6 have a high sensitivity7 ; however, only a subset of anti-PF4/heparin antibodies that activate platelets is clinically relevant. In some patient populations, such as patients undergoing cardiac surgery, this accounts for <25% of anti-PF4/heparin antibodies detected by antigen tests.8 Clinically relevant anti-PF4/heparin antibodies typically activate washed platelets of healthy donors in the presence of patient serum and heparin in the heparin-induced platelet activation (HIPA) test9 or the serotonin release assay.10,11
Platelet activation by HIT antibodies is caused via the platelet FcγRIIA receptor and depends on cosignaling via the P2Y12 ADP receptor,12 which is blocked by ticagrelor. When we observed a patient with a clinical course typical for HIT, a positive anti-PF4/heparin enzyme immunoassay (EIA) but a negative HIPA test (Figure 1), we systematically assessed whether ticagrelor may lead to a false-negative HIPA test. The in-house anti-PF4/heparin-immunoglobulin G (IgG) EIA and HIPA tests were performed as previously described.9,13,14 Aggregation in ≥3 of 4 donors’ test cells in the presence of low molecular weight heparin (0.2 aFXaU/mL) within 35 minutes was defined as HIT+. Confirmed HIT+ sera were spiked with ticagrelor (final concentrations: 15.6, 31.3, 62.5, 125, 250, 500, and 1000 ng/mL; in vivo range, 227-770 ng/mL; 1% dimethyl sulfoxide was used as carrier control). The IgG fraction from patient sera was prepared using protein G sepharose (optical density [OD] 2.20 in the PF4/heparin EIA). Serum from the index patient (Figure 1) and ticagrelor-spiked known HIPA+ sera were incubated for 30 minutes with 100 μg/mL the ticagrelor antidote PB2452 (kindly provided by PhaseBio, Malvern, PA). A total of 500 μL of HIPA+ sera spiked with ticagrelor was incubated with 20 mg of pulverized activated charcoal tablets (Kohle-Hevert, Hevert Arzneimittel, Berlin, Germany) for 5 minutes, then centrifuged, and the supernatant was used.
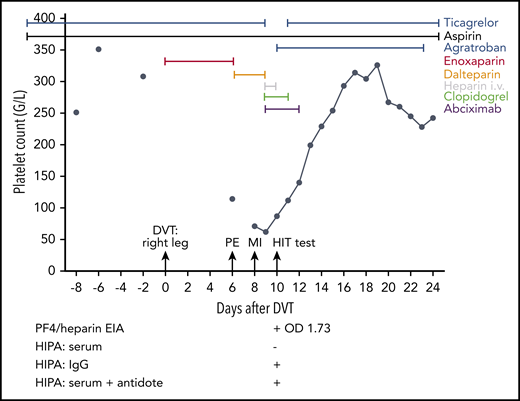
At day −20, the 59-year-old female patient had an ST-segment elevation myocardial infarction and cardiac arrest. She required cardiopulmonary resuscitation during which a bolus of unfractionated heparin was given, followed by placement of 3 drug-eluting stents in the right coronary artery. Resuscitation was complicated by liver laceration requiring surgery. No thrombosis prophylaxis was given because of the anticipated risk of bleeding after liver injury. Therapeutic-dose low molecular weight heparin was started at day 0 upon diagnosis of a deep vein thrombosis (DVT) of the right leg (enoxaparin; 1 mg/kg body weight twice a day subcutaneously). Severe pulmonary embolism (PE) occurred at day 6, and the platelet count had decreased to 114 per microliter. Anticoagulation was switched to dalteparin (5000 aFXaU twice a day subcutaneously). Three days later, the patient was transferred to the hospital of 1 of the authors (C.P.) with a further myocardial infarction (MI) due to in-stent thrombosis of the right coronary artery. Heparin-induced thrombocytopenia was suspected; the 4T score was 7. All heparins were stopped, and anticoagulation was switched to argatroban. In addition, abciximab and clopidogrel were given. The patient was discharged at day 25 and has remained stable. “HIT test” denotes the time point at which a blood sample was taken for HIT diagnosis; test results are shown below the graph. The PF4/heparin EIA was strongly positive (OD 1.73), but the HIPA was negative.
At day −20, the 59-year-old female patient had an ST-segment elevation myocardial infarction and cardiac arrest. She required cardiopulmonary resuscitation during which a bolus of unfractionated heparin was given, followed by placement of 3 drug-eluting stents in the right coronary artery. Resuscitation was complicated by liver laceration requiring surgery. No thrombosis prophylaxis was given because of the anticipated risk of bleeding after liver injury. Therapeutic-dose low molecular weight heparin was started at day 0 upon diagnosis of a deep vein thrombosis (DVT) of the right leg (enoxaparin; 1 mg/kg body weight twice a day subcutaneously). Severe pulmonary embolism (PE) occurred at day 6, and the platelet count had decreased to 114 per microliter. Anticoagulation was switched to dalteparin (5000 aFXaU twice a day subcutaneously). Three days later, the patient was transferred to the hospital of 1 of the authors (C.P.) with a further myocardial infarction (MI) due to in-stent thrombosis of the right coronary artery. Heparin-induced thrombocytopenia was suspected; the 4T score was 7. All heparins were stopped, and anticoagulation was switched to argatroban. In addition, abciximab and clopidogrel were given. The patient was discharged at day 25 and has remained stable. “HIT test” denotes the time point at which a blood sample was taken for HIT diagnosis; test results are shown below the graph. The PF4/heparin EIA was strongly positive (OD 1.73), but the HIPA was negative.
In all but 1 tested sera, ticagrelor dose dependently inhibited the HIPA test when concentrations in the serum reached those during clinical steady-state (227-770 ng/mL; Figure 2A). In contrast to the ticagrelor-containing serum, the purified IgG fraction (PF4/heparin EIA OD 2.20) strongly activated platelets in the HIPA test (Figure 2A,B); respiking with ticagrelor again inhibited the HIPA test. Also in the presence of the ticagrelor antidote (PB2452; 100 mg/mL), consisting of monoclonal Fab fragments against ticagrelor that block ticagrelor-platelet interactions,15 the index patient’s serum and ticagrelor (500 ng/mL)–containing patient sera tested positive in the HIPA test (Figure 2C). Finally, when we incubated 3 previously confirmed HIPA+ sera, prespiked with ticagrelor (500 ng/mL) with activated charcoal, the cleared sera again tested positive in the HIPA test (Figure 2D). The Institutional Review Board of the Universitätsmedizin Greifswald, Greifswald, Germany, allowed the use of anonymized patient samples for laboratory studies. The patient described in the case report gave informed consent for publishing the case report.
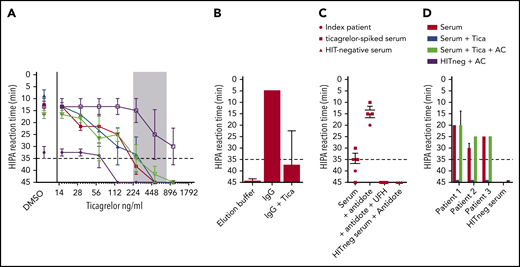
Ticagrelor inhibits platelet activation by HIT antibodies in functional assays. (A) Five different known HIT+ sera were spiked with increasing concentrations of ticagrelor and assessed using the HIPA test in the presence of 0.2 aFXaU/mL reviparin, with dimethyl sulfoxide as a carrier control. The lag time until platelet aggregation is shown on the y-axis (reversed). A lag time < 35 minutes (dotted line) corresponds to a positive HIPA test. The ticagrelor concentration used is shown on the x-axis, with the observed therapeutic range in patients depicted as a shaded area. Four of 5 tested sera became negative when ticagrelor was present in this range. Error bars represent the range of lag time observed in 4 donor test cells. (B) Total IgG was isolated from the index patient’s serum and tested using the HIPA test, which resulted in fast and strong activation of donor platelets (within 5 minutes). When the IgG fraction was respiked with ticagrelor, the HIPA test was again inhibited. The elution buffer served as a negative control. Data are shown as the mean and standard deviation (SD) of lag time observed with washed platelets from 3 donors. (C) Two sera (ie, the index patient and a serum known to contain platelet-activating antibodies and prespiked with ticagrelor) were tested in the HIPA test in the presence or absence of the ticagrelor antidote PB2452. In the absence of PB2452, both sera gave false-negative results, which were fully reversed by PB2452. Inhibition by high-dose heparin confirmed the specificity of the reaction and ruled out an unspecific effect of PB2452; PB2452 does not activate platelets in the case of HIT− serum. Data are shown as mean and SD of lag time of the 2 tested sera with washed platelets of 3 donors. (D) Sera of 3 known HIT patients were tested in the HIPA test. For all 3 HIT cases, the sera were positive in the presence of heparin and became negative after being spiked with ticagrelor. When the ticagrelor-spiked serum was also incubated with activated charcoal (AC), the HIPA test was positive again. No unspecific activation of platelets by AC was observed (HIT− serum incubated with AC). Data are shown as mean and SD of lag time of the tested sera with ≥3 donors. AC, 20 mg of active charcoal powder; Tica, ticagrelor; UFH, high-dose heparin (100 IU/mL).
Ticagrelor inhibits platelet activation by HIT antibodies in functional assays. (A) Five different known HIT+ sera were spiked with increasing concentrations of ticagrelor and assessed using the HIPA test in the presence of 0.2 aFXaU/mL reviparin, with dimethyl sulfoxide as a carrier control. The lag time until platelet aggregation is shown on the y-axis (reversed). A lag time < 35 minutes (dotted line) corresponds to a positive HIPA test. The ticagrelor concentration used is shown on the x-axis, with the observed therapeutic range in patients depicted as a shaded area. Four of 5 tested sera became negative when ticagrelor was present in this range. Error bars represent the range of lag time observed in 4 donor test cells. (B) Total IgG was isolated from the index patient’s serum and tested using the HIPA test, which resulted in fast and strong activation of donor platelets (within 5 minutes). When the IgG fraction was respiked with ticagrelor, the HIPA test was again inhibited. The elution buffer served as a negative control. Data are shown as the mean and standard deviation (SD) of lag time observed with washed platelets from 3 donors. (C) Two sera (ie, the index patient and a serum known to contain platelet-activating antibodies and prespiked with ticagrelor) were tested in the HIPA test in the presence or absence of the ticagrelor antidote PB2452. In the absence of PB2452, both sera gave false-negative results, which were fully reversed by PB2452. Inhibition by high-dose heparin confirmed the specificity of the reaction and ruled out an unspecific effect of PB2452; PB2452 does not activate platelets in the case of HIT− serum. Data are shown as mean and SD of lag time of the 2 tested sera with washed platelets of 3 donors. (D) Sera of 3 known HIT patients were tested in the HIPA test. For all 3 HIT cases, the sera were positive in the presence of heparin and became negative after being spiked with ticagrelor. When the ticagrelor-spiked serum was also incubated with activated charcoal (AC), the HIPA test was positive again. No unspecific activation of platelets by AC was observed (HIT− serum incubated with AC). Data are shown as mean and SD of lag time of the tested sera with ≥3 donors. AC, 20 mg of active charcoal powder; Tica, ticagrelor; UFH, high-dose heparin (100 IU/mL).
These data show that ticagrelor present in patient’s serum during treatment with ticagrelor inhibits the functional assay for HIT. This inhibition can be ameliorated by 3 methods: isolating the IgG fraction from the patient serum, preincubating the serum with the ticagrelor antidote PB2452, or preincubating the patient serum with 20 mg of charcoal powder.
This finding is of major relevance when patients suspected to have HIT also receive ticagrelor. Although it is not very frequent, HIT can occur in cardiac patients with acute coronary syndrome, as in the index case presented. Cardiac patients with additional major complications are at an especially increased risk for HIT. In the index case, the tissue trauma after resuscitation was probably an additional trigger for the development of HIT. In contrast to medically treated cardiac patients, HIT is highly relevant in patients undergoing cardiac surgery. The incidence of HIT after cardiac surgery is in the range of 1% to 2%,16 and it is ∼0.5% after transcatheter aortic valve replacement.17 Although ticagrelor is usually stopped before surgery, antiplatelet drugs are typically restarted within 3 days after major surgery in patients at high risk for coronary events, whereas HIT typically occurs 5 to 10 days after major surgery. Thus, restart of ticagrelor and the onset of HIT after surgery will usually overlap. Although ticagrelor in the patient’s serum will inhibit the in vitro test, the index patient demonstrates that venous and arterial thrombotic complications can be induced by heparin in acute HIT in vivo, despite concomitant ticagrelor treatment.
An additional clinically important scenario is patients with a (recent) history of HIT who require cardiac surgery. Heparin is still the most preferred anticoagulant during cardiopulmonary bypass surgery. In patients with a history of HIT, who require cardiopulmonary bypass surgery with the heart-lung machine, current guidelines recommend the use of heparin once the functional test becomes negative, even when the antigen test is still positive (and to use nonheparin anticoagulants before and after surgery).7
As a result of the widespread use of ticagrelor, the number of patients in whom HIT is suspected while they are treated with ticagrelor will likely increase. Continuing or restarting heparin based on a false-negative platelet-activation test can be life-threatening. For interpretation of platelet-activation tests, the laboratory needs to know whether the patient is treated with ticagrelor. Removing ticagrelor from the patient’s serum by charcoal powder, using the purified IgG fraction, or inhibiting ticagrelor in the patient’s serum by a specific antidote is a method to obtain reliable results in functional HIT tests.
Although we did not test this in the present study, it is very likely that the other available reversibly binding P2Y12 inhibitor, cangrelor, shows the same effect. However, because of its short half-life, this can be overcome by stopping the drug up to 30 minutes before the blood sample for HIT testing is obtained. We cannot exclude that prasugrel and clopidogrel also show some inhibition at peak plasma concentrations; however, their plasma concentrations are much lower than for ticagrelor. Furthermore, the active metabolites are highly unstable in serum because they contain a thiol group. Because the timespan between taking a blood sample and testing the serum in a functional test is several hours, and the serum is heat inactivated, we consider it highly unlikely that the unstable metabolite of clopidogrel will still be present in the serum at sufficient concentrations.
In summary, free P2Y12 inhibitors in patient plasma can inhibit functional tests for HIT. This is of special importance for the testing of cardiac patients with a history of (recent) HIT who are scheduled for planned re-exposure to heparin during cardiopulmonary bypass surgery. It is very important that clinicians inform the laboratory about the use of ticagrelor in a patient when the clinical suspicion for HIT should be confirmed or ruled out. Laboratory physicians must be aware of the interaction between ticagrelor and functional assays for HIT.
Acknowledgments
The authors thank Ulrike Strobel and Ricarda Raschke for technical support.
This work was supported by Deutsche Forschungsgemeinschaft Grant SFBTRR240: Z03.
Authorship
Contribution: J.J.M.E. performed ticagrelor experiments and wrote the manuscript; C.P., T.M., and G.W. managed the patient with HIT under ticagrelor, first recognized that the functional HIT test was likely false negative, and wrote the patient case report; N.K. performed all charcoal experiments; A.G. designed the study, interpreted the results, and wrote the manuscript; and all authors reviewed the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Andreas Greinacher, Institut für Immunologie und Transfusionsmedizin, Universitätsmedizin Greifswald, Sauerbruchstraße, 17487 Greifswald, Germany; e-mail: andreas.greinacher@med.uni-greifswald.de.

