

TO THE EDITOR:
The impact of mutations in DNA methyltransferase 3 α (DNMT3A) at diagnosis as a prognostic marker in acute myeloid leukemia (AML) has been contradictory so far.1-3 These discrepancies most likely arise from differences of therapeutic protocols used. Most important, few if any studies have evaluated the clinical importance of the 3-way co-occurrence of mutations affecting DNMT3A, nucleophosmin (NPM1), and fms-like tyrosine kinase 3 (FLT3) genes (in particular FLT3 length mutations or FLT3–internal tandem duplication (ITD) for “internal tandem duplication”) in patients treated outside of well-controlled clinical trials, a real-life setting that represents most low- and middle-income countries. Hence, we assessed the frequency and clinical impact of DNMT3A mutations and the co-occurrence of DNMT3A/NPM1/FLT3-ITD mutations on treatment outcomes of nonselected AML patients, followed from June 2003 to January 2019 at 5 Brazilian reference centers specialized on AML treatment.
Bone marrow samples from 507 consecutive patients with de novo AML (median age, 51 years; range, 18-94 years; 47% male) were obtained at diagnosis. Patients with acute promyelocytic leukemia, therapy-related AML, or with a previous history of myelodysplastic syndrome were excluded. Details for treatment protocols can be found in the supplemental Methods, available on the Blood Web site. The study adhered to the tenets of the Declaration of Helsinki and informed consents were obtained from all patients or their relatives. The local Research Ethics Board of each participating center approved the study.
The DNMT3A and NPM1 mutations were analyzed by standard sequencing techniques. Details are described in supplemental Methods. Screening for FLT3-ITD mutations was performed by polymerase chain reaction according to the method of Kiyoi et al.4 In parallel, we explored the FLT3 allelic ratio in patients with the FLT3-ITD mutated status. FLT3-tyrosine kinase domain mutations were not evaluated in this study. Because most of the DNMT3A mutations in myeloid neoplasms occur at exon 23, with a significant enrichment for mutations at codon R882,5,6 we evaluated the mutational and phenotypic profile of patients harboring DNMT3A-R882 and non-R882 mutations using The Cancer Genome Atlas database (TCGA) data set.5 We observed that NPM1 and FLT3-ITD mutations were significantly enriched in DNMT3A-R882 when compared with non-R882 mutations or DNMT3A wild type (supplemental Figure 1). Therefore, based on its biological and clinical significance3,7-10 and our own experience, screening for DNMT3A mutations was restricted to the codon R882.
DNMT3A-R882 mutations were detected in 64 of 507 patients (13%), most of them identified as R882H (49 of 64; 76%), followed by R882C (12 of 64; 19%), and R882P (3 of 64; 5%). Samples without detectable DNMT3A-R882 mutations or carrying single-nucleotide polymorphisms are referenced here as “DNMT3A nonmutated.” To decide which variables to include in the multivariate Cox proportional hazard model, we performed a backward elimination analysis using the Akaike Information Criteria (AIC) as fitness measure and getting the best-fitted model (supplemental Table 1). The basis (indispensable) variable used was cytogenetic risk stratification. Treatment-related variables were not included in the multivariate model due to the biased nature of a retrospective study.
The clinical and baseline characteristics are summarized in Table 1. Overall, 302 of 507 patients (60%) achieved complete remission (CR), of whom 37 of 64 (53%) and 265 of 443 (60%) were assigned to the DNMT3A-mutated and DNMT3A-nonmutated groups, respectively (P = .786). The median follow-up among survivals was 39 months (95% confidence interval [CI], 26-53 months). Patients with DNMT3A mutations had significantly lower 5-year overall survival (OS) (9%; 95% CI, 3% to 18%) compared with those without DNMT3A mutations (22%; 95% CI, 17% to 27%) (P = .0035) (Figure 1A). The best-fitted multivariate Cox proportional hazards model for OS was age (>60 years old), leukocyte counts (>50 × 109/L), DNMT3A status, and cytogenetic risk stratification. This model showed that DNMT3A mutational status was independently associated with poor OS (hazard ratio [HR], 1.4; 95% CI, 1.01-2.1; P = .04) (Figure 1G). Of the 302 patients who achieved CR, 133 patients (44%) relapsed. Considering nonrelapse death as a competing cause of failure, the 5-year cumulative incidence of relapse (CIR) rate was 54% (95% CI, 48% to 60%). CIR rates for patients assigned to the DNMT3A mutated and nonmutated groups were 72% (95% CI, 58% to 86%) and 50% (95% CI, 42% to 57%), respectively (P < .0001; Figure 1B). Patients with DNMT3A mutations had a significantly lower disease-free survival (DFS) rate (19%; 95% CI, 7% to 34%) in comparison with patients without DNMT3A mutations (42%; 95% CI, 34% to 49%) (P < .0001; Figure 1C).
Clinical and baseline characteristics according to the DNMT3A mutational status and according to the DNMT3A/NPM1/FLT3-ITD mutations
| Characteristics | All patients, no. (%) | DNMT3A mutated, no. (%) | DNMT3A nonmutated, no. (%) | P* | Triple-mutated, no. (%) | Non–triple-mutated, no. (%) | P* |
|---|---|---|---|---|---|---|---|
| Age, y | <.001† | .003† | |||||
| 18-40 | 157 (31) | 6 (9.4) | 151 (34.1) | 3 (8.6) | 154 (32.6) | ||
| 41-60 | 185 (36.5) | 34 (53.1) | 151 (34.1) | 21 (60) | 164 (34.7) | ||
| 60 and older | 165 (32.5) | 24 (37.5) | 141 (31.8) | 11 (31.4) | 154 (32.6) | ||
| Median (range) | 50.6 (18, 93.8) | 54.4 (27, 91) | 49.2 (18, 93.8) | .003† | 54.8 (27, 77.5) | 49.9 (18, 93.8) | .08 |
| Sex | .033† | .292 | |||||
| Female | 269 (53.1) | 42 (66.6) | 227 (51.2) | 22 (62.9) | 247 (52.3) | ||
| Male | 238 (46.9) | 22 (34.4) | 216 (48.8) | 13 (37.1) | 225 (47.7) | ||
| FAB subtype | .756 | .928 | |||||
| M0 | 22 (4.7) | 2 (3.4) | 20 (4.9) | 1 (3.1) | 21 (4.8) | ||
| M1 | 90 (19.3) | 9 (15.3) | 81 (19.9) | 7 (21.9) | 83 (19.1) | ||
| M2 | 150 (32.1) | 17 (28.8) | 133 (32.6) | 12 (37.5) | 138 (31.7) | ||
| M4 | 150 (32.1) | 23 (39) | 127 (31.1) | 10 (31.3) | 140 (32.2) | ||
| M5 | 41 (8.8) | 7 (11.9) | 34 (8.3) | 2 | 39 (9) | ||
| M6 | 10 (2.1) | 1 (1.7) | 9 (2.2) | 10 (2.3) | |||
| M7 | 4 (0.9) | 4 (1) | 4 (0.9) | ||||
| Missing data | 40 | 5 | 35 | 3 | 37 | ||
| Cytogenetic risk stratification‡ | <.001† | .006† | |||||
| Favorable | 69 (19.5) | 69 (22.6) | 69 (21.1) | ||||
| Intermediate | 227 (64.3) | 43 (89.6) | 184 (60.3) | 24 (92.3) | 203 (62.1) | ||
| Adverse | 57 (16.1) | 5 (10.4) | 52 (17) | 2 (7.7) | 55 (16.8) | ||
| Missing data§ | 154 | 16 | 138 | 145 | |||
| FLT3-ITD | <.001† | ||||||
| Mutated | 134 (26.4) | 42 (65.6) | 94 (20.8) | ||||
| Nonmutated | 373 (73.1) | 22 (34.4) | 351 (79.2) | ||||
| NPM1 | <.001† | ||||||
| Mutated | 145 (28.6) | 44 (68.8) | 103 (22.8) | ||||
| Nonmutated | 362 (71.4) | 20 (31.3) | 342 (77.2) | ||||
| BM blasts, median (range), % | 67 (20, 100) | 74 (20, 96) | 66 (10, 100) | .143 | 77 (21, 96) | 66 (20, 100) | .117 |
| Leukocyte count, median (range), ×109/L | 27.3 (0.28, 790) | 61 (0.28, 435) | 25.6 (0.6, 790) | .009† | 67 (0.86, 435) | 26 (0.28, 790) | .001† |
| Platelet count, median (range), ×109/L | 45 (3, 600) | 59 (5, 404) | 44 (3, 600) | .818 | 60 (10, 196) | 45 (3, 600) | .763 |
| Hg, median (range), g/dL | 8.1 (3, 16.2) | 8 (3.1, 11.6) | 8.1 (3, 16.2) | .727 | 7.8 (3.9, 13.2) | 8.1 (3, 16.2) | .534 |
| LDH level, median (range), U/I | 682.5 (116, 11 722) | 775 (136, 5289) | 662 (116, 11 722) | .188 | 767 (136, 2789) | 672 (116, 11 722) | .607 |
| Complete remission, % | 60 | 53 | 60 | .786 | 62 | 59 | .725 |
| OS, % (95% CI) | 20 (16, 24) | 9 (3, 18) | 22 (17, 27) | .003† | 3 (0-14) | 22 (17, 26) | .011† |
| DFS, % (95% CI) | 38 (31, 45) | 19 (7, 34) | 42 (34, 49) | <.001† | 4 (0-19) | 42 (35, 50) | <.001† |
| CIR, % (95% CI) | 54 (48, 60) | 72 (58, 86) | 50 (42, 57) | <.001† | 85 (71, 98) | 50 (43, 56) | <.001† |
| Characteristics | All patients, no. (%) | DNMT3A mutated, no. (%) | DNMT3A nonmutated, no. (%) | P* | Triple-mutated, no. (%) | Non–triple-mutated, no. (%) | P* |
|---|---|---|---|---|---|---|---|
| Age, y | <.001† | .003† | |||||
| 18-40 | 157 (31) | 6 (9.4) | 151 (34.1) | 3 (8.6) | 154 (32.6) | ||
| 41-60 | 185 (36.5) | 34 (53.1) | 151 (34.1) | 21 (60) | 164 (34.7) | ||
| 60 and older | 165 (32.5) | 24 (37.5) | 141 (31.8) | 11 (31.4) | 154 (32.6) | ||
| Median (range) | 50.6 (18, 93.8) | 54.4 (27, 91) | 49.2 (18, 93.8) | .003† | 54.8 (27, 77.5) | 49.9 (18, 93.8) | .08 |
| Sex | .033† | .292 | |||||
| Female | 269 (53.1) | 42 (66.6) | 227 (51.2) | 22 (62.9) | 247 (52.3) | ||
| Male | 238 (46.9) | 22 (34.4) | 216 (48.8) | 13 (37.1) | 225 (47.7) | ||
| FAB subtype | .756 | .928 | |||||
| M0 | 22 (4.7) | 2 (3.4) | 20 (4.9) | 1 (3.1) | 21 (4.8) | ||
| M1 | 90 (19.3) | 9 (15.3) | 81 (19.9) | 7 (21.9) | 83 (19.1) | ||
| M2 | 150 (32.1) | 17 (28.8) | 133 (32.6) | 12 (37.5) | 138 (31.7) | ||
| M4 | 150 (32.1) | 23 (39) | 127 (31.1) | 10 (31.3) | 140 (32.2) | ||
| M5 | 41 (8.8) | 7 (11.9) | 34 (8.3) | 2 | 39 (9) | ||
| M6 | 10 (2.1) | 1 (1.7) | 9 (2.2) | 10 (2.3) | |||
| M7 | 4 (0.9) | 4 (1) | 4 (0.9) | ||||
| Missing data | 40 | 5 | 35 | 3 | 37 | ||
| Cytogenetic risk stratification‡ | <.001† | .006† | |||||
| Favorable | 69 (19.5) | 69 (22.6) | 69 (21.1) | ||||
| Intermediate | 227 (64.3) | 43 (89.6) | 184 (60.3) | 24 (92.3) | 203 (62.1) | ||
| Adverse | 57 (16.1) | 5 (10.4) | 52 (17) | 2 (7.7) | 55 (16.8) | ||
| Missing data§ | 154 | 16 | 138 | 145 | |||
| FLT3-ITD | <.001† | ||||||
| Mutated | 134 (26.4) | 42 (65.6) | 94 (20.8) | ||||
| Nonmutated | 373 (73.1) | 22 (34.4) | 351 (79.2) | ||||
| NPM1 | <.001† | ||||||
| Mutated | 145 (28.6) | 44 (68.8) | 103 (22.8) | ||||
| Nonmutated | 362 (71.4) | 20 (31.3) | 342 (77.2) | ||||
| BM blasts, median (range), % | 67 (20, 100) | 74 (20, 96) | 66 (10, 100) | .143 | 77 (21, 96) | 66 (20, 100) | .117 |
| Leukocyte count, median (range), ×109/L | 27.3 (0.28, 790) | 61 (0.28, 435) | 25.6 (0.6, 790) | .009† | 67 (0.86, 435) | 26 (0.28, 790) | .001† |
| Platelet count, median (range), ×109/L | 45 (3, 600) | 59 (5, 404) | 44 (3, 600) | .818 | 60 (10, 196) | 45 (3, 600) | .763 |
| Hg, median (range), g/dL | 8.1 (3, 16.2) | 8 (3.1, 11.6) | 8.1 (3, 16.2) | .727 | 7.8 (3.9, 13.2) | 8.1 (3, 16.2) | .534 |
| LDH level, median (range), U/I | 682.5 (116, 11 722) | 775 (136, 5289) | 662 (116, 11 722) | .188 | 767 (136, 2789) | 672 (116, 11 722) | .607 |
| Complete remission, % | 60 | 53 | 60 | .786 | 62 | 59 | .725 |
| OS, % (95% CI) | 20 (16, 24) | 9 (3, 18) | 22 (17, 27) | .003† | 3 (0-14) | 22 (17, 26) | .011† |
| DFS, % (95% CI) | 38 (31, 45) | 19 (7, 34) | 42 (34, 49) | <.001† | 4 (0-19) | 42 (35, 50) | <.001† |
| CIR, % (95% CI) | 54 (48, 60) | 72 (58, 86) | 50 (42, 57) | <.001† | 85 (71, 98) | 50 (43, 56) | <.001† |
Values are shown as number (percentage), unless otherwise specified in the row headings as median (range) or percentage (95% CI).
BM, bone marrow; CIR, cumulative incidence of relapse; DFS, disease-free survival; FAB, French-American-British; Hg, hemoglobin; LDH, lactate dehydrogenase; OS, overall survival
Missing values were excluded for the calculation of P values.
Indicates statistically significant differences.
The cytogenetic risk groups were defined according to Medical Research Council criteria.24
Material not available or no metaphases detected.
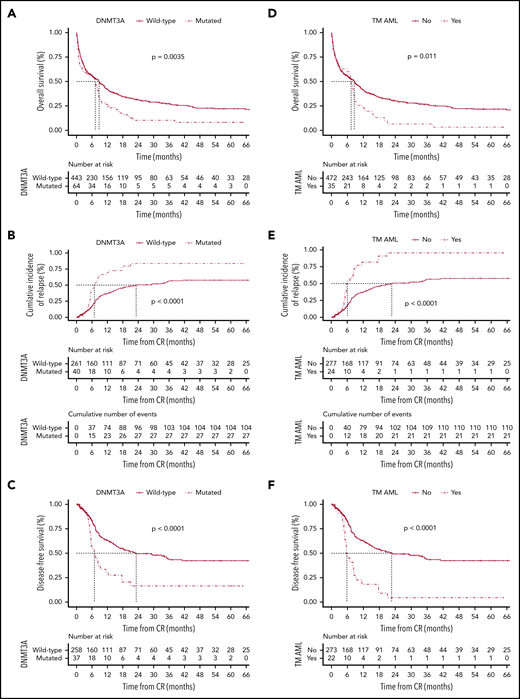

Probability according to DNMT3A mutations. The probability of overall survival (A), cumulative incidence of relapse (B), and disease-free survival (C) in patients with AML according to DNMT3A mutations. Overall survival (D), cumulative incidence of relapse (E), and disease-free survival (F) in triple-mutated (TM-AML) patients. Survival curves were estimated using the Kaplan-Meier method, and the log‐rank test was used for comparison. Cumulative incidence curves for nonrelapse death and relapse with or without death were constructed to reflect time to relapse and time to nonrelapse death as competing risks. Time to relapse and time to nonrelapse death were measured from the date of complete remission. Multivariate Cox model for overall survival (G) and disease-free survival (H).
Probability according to DNMT3A mutations. The probability of overall survival (A), cumulative incidence of relapse (B), and disease-free survival (C) in patients with AML according to DNMT3A mutations. Overall survival (D), cumulative incidence of relapse (E), and disease-free survival (F) in triple-mutated (TM-AML) patients. Survival curves were estimated using the Kaplan-Meier method, and the log‐rank test was used for comparison. Cumulative incidence curves for nonrelapse death and relapse with or without death were constructed to reflect time to relapse and time to nonrelapse death as competing risks. Time to relapse and time to nonrelapse death were measured from the date of complete remission. Multivariate Cox model for overall survival (G) and disease-free survival (H).
Considering its clinical11 and biological9,10,12 relevance and the strikingly high frequency of 3-way co-occurrence mutations in AML,5 we evaluated the clinical relevance of the co-occurrence DNMT3A/NPM1/FLT3-ITD in a real-life setting. The frequency of triple-mutated patients in our cohort (35 of 507; 7%) was very similar to other studies.5,11 Table 1 summarized the main baseline and clinical characteristics. Triple-mutated patients had significantly lower OS (4%; 95% CI, 2% to 15% vs 21%, 95% CI, 17% to 26%; P = .011), higher CIR rate (85%; 95% CI, 71% to 98% vs 50%, 95% CI, 43% to 56%; P < .0001), and lower DFS rates (5%; 95% CI, 1% to 20% vs 42%, 95% CI, 34% to 49%; P < .0001) compared with non–triple-mutated patients (Figure 1D-F). In multivariate analysis, the lowest AIC for DFS was achieved when FLT3-ITD and NPM1 status, triple-mutant AML, and cytogenetic risk stratification were included. In this model, triple-mutant AML presented a higher relapse risk (HR, 2.49, 95% CI, 1.3-5.5; P = .02) (Figure 1H). The co-occurrence of DNMT3A/NPM1/FLT3-ITD mutations had no impact on CR achievement (P = .725).
To validate our findings, we took advantage of 2 publicly available AML data sets (TCGA5 and Gene Expression Omnibus,13 GEO; www.ncbi.nlm.nih.gov/geo, accession number: GSE6891). For survival analysis, only de novo AML patients submitted to intensive therapy were included. DNMT3A-R882 mutations were reported in 22 of 142 (15%) and 64 of 479 patients (13%) included in the TCGA and GEO cohorts, respectively. DNMT3A mutations were significantly associated with poor OS (HR, 1.65; 95% CI, 1.1-2.7; P = .046), but not with DFS (HR, 1.64; 95% CI, 0.95-2.83; P = .072) in TCGA patients. In contrast, DNMT3A mutational status had no impact on OS (HR, 1.22; 95% CI, 0.95-1.58; P = .115), but was significantly associated with poor DFS for patients included in the GEO cohort (HR, 1.47; 95% CI, 1.1-2; P = .015). The co-occurrence of DNMT3A/NPM1/FLT3-ITD mutations was significantly associated with poor DFS (HR, 1.3, 95% CI, 1.1-1.68; P = .038), but not with OS (HR, 1.16, 95% CI, 0.92-1.48; P = .19) in TCGA patients. Similar results were obtained for GEO patients: DFS (HR, 2.02; 95% CI, 1.3-3.17; P = .002) and OS (HR, 1.5; 95% CI, 1.01-2.24; P = .48).
In summary, we demonstrated that DNMT3A mutations might be useful for AML outcome prediction, although results remain conflicting. Several groups speculate that patient-related features and differences in treatment protocols could explain the contradictory results.1-3,7 In fact, the functional genomic landscape of AML suggest that the response to drugs is specific to combinatorial mutational events.14 Therefore, the impact of DNMT3A mutations in clinical decision-making remains disputable. Importantly, we restricted the screening for DNMT3A mutations to codon R882, which probably explains our lower rate of DNMT3A mutations in comparison with other studies.1,2 Although, one may argue that restricting our analysis to the codon R882 may limit our study, it is important to notice that current literature supports the idea that only DNMT3A-R882 mutations contribute to prognostication3,7,15 and pathophysiology of AML.9,10,16 For instance, non-R882 mutations found on the DNMT3A enzyme affecting different domains showed few biochemical consequences.17 Furthermore, DNMT3A-R882 mutations (but not non-R882 mutations) may have an impact on clonal hematopoiesis.18-20 Finally, a remarkable difference in DNA methylation signatures between samples with DNMT3A-R882 and non-R882 mutations has been reported,8 suggesting that these mutations should not be pooled together. It is possible that mutations in different DNMT3A domains lead to different neomorphic functions, resulting in pathogenetic variabilities. Nevertheless, whether DNMT3A non-R882 mutations harbor biological or clinical importance in AML requires further studies.
Yet, the co-occurrence DNMT3A/NPM1/FLT3-ITD mutations is more representative regarding the biology of the disease and may constitute a more robust strategy for outcome prediction in AML. Reasons for such robustness remain to be elucidated, although functional studies have identified a link between the co-occurrence of DNMT3A/NPM1/FLT3-ITD mutations and AML resistance to anthracycline based-chemotherapy.9 In agreement, triple-mutated AML showed a unique differentiation response to the FLT3 inhibitor AC22021 and increased sensitivity to the Food and Drug Administration-approved drug ibrutinib.14 More recently, transcriptomic and immunophenotypic data describe triple-mutant blasts to be associated with high leukemia stem cell frequency, and synergistic upregulation of specific leukemia stem cell regulator.12 Finally, specific DNA methylation signatures were characterized in triple-mutated patients.22 We speculate that these findings may help us to better understand the poor prognosis of this specific AML subtype.
This is one of the first studies to describe the prognostic importance of DNMT3A mutations and the co-occurrence of DNMT3A/NPM1/FLT3-ITD mutations involving consecutive nonselected patients treated outside well-controlled clinical trials. As such, any conclusion drawn from this “real-world” data should be interpreted with caution. In contrast to previous studies that draw their conclusions based on a uniformly treated patient population,11 our study is confronted with many variables (including drug unavailability, risk-adapted treatment, comorbidities and time from diagnosis to treatment initiation) that cannot be fully controlled. Nevertheless, we are firm believers that clinical data obtained from real-world studies, if properly registered and with guaranteed accessibility, can provide representative evidence from routine practice about the clinical outcomes of patients, without the classical selection criteria of clinical trials.23 Most importantly, these findings can serve as a more reliable basis for extrapolation of data to understudied populations.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Erich V. de Paula for intellectual support and critical revision of this manuscript.
This work was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; grant #444656/2014-2016). M.F.B. received a fellowship from Fundação de Amparo a Ciência e Tecnologia de Pernambuco (grant #ibpg-0448-2.02/14).
Authorship
Contribution: M.F.B. performed experiments, collected, analyzed, and interpreted data, and drafted the manuscript; D.R.S. and A.R.L.-A. performed the statistical analyses, interpreted the data, and drafted the manuscript; A.S.L., M.-R.P.-B., D.R.S., J.L.C.-S., D.A.P.-M., I.W., P.L.F.-N., L.Q., A.C., M.M.O., M.M.L., R.A.d.A., P.d.M.C., B.K.D., I.B., V.R., E.M.R., F.T., S.T.S., E.I.B., and M.A.B. obtained patient samples, updated the clinical data, collected, analyzed, and interpreted data, and drafted the manuscript; M.F.B. and A.R.L.-A. conceived and designed the study and reviewed the manuscript; and A.R.L.-A. gave final approval of the version to be submitted.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Antonio R. Lucena-Araujo, Department of Genetics, Federal University of Pernambuco, Av. Prof. Moraes Rego, 1235, Recife PE 50670-901, Brazil; e-mail: araujoarl@gmail.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal