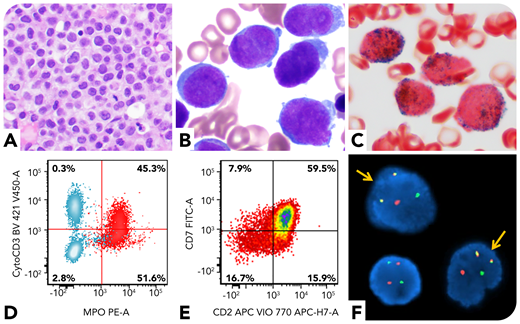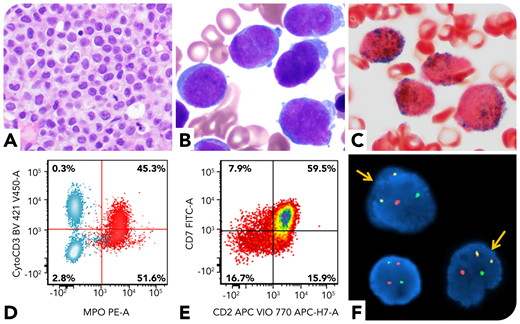
A 59-year-old man presented with oral bleeding. Peripheral blood showed leukocytosis (59.6 × 109/L; 83% blasts). Bone marrow biopsy revealed markedly increased immature cells (panel A; hematoxylin and eosin stain, ×40 objective, original magnification ×400). Aspirate smear showed blasts with round to irregular nuclei, fine chromatin, and small amount of cytoplasm; no obvious Auer rods were identified (panel B; Wright and Giemsa stains, ×100 objective, original magnification ×1000). The blasts were positive for myeloperoxidase (panel C; cytochemical stain, ×100 objective, original magnification ×1000). Flow cytometry showed the blasts (CD34+CD117+HLA-DR−) coexpressed myeloid (myeloperoxidase, CD13, and CD33) and T-lineage (cytoplasmic CD3, CD2, and CD7) markers, suggestive of mixed-phenotype acute leukemia (MPAL), T/myeloid (panels D-E; red cells in panel D are blasts, and blue cells in panel D are T and B cells in the background). However, his D-dimer level was markedly increased (>20 μg/mL fibrinogen equivalent units; normal range 0-0.4), accompanied by a decreased fibrinogen level, concerning for acute promyelocytic leukemia (APL). Fluorescence in situ hybridization confirmed PML/RARA rearrangement (panel F, yellow arrows; original magnification ×1000). Molecular studies revealed an internal tandem duplication and a D835 point mutation of the FLT3 gene. He was diagnosed with microgranular APL.
APL is a hematologic emergency due to its high mortality rate. Cytoplasmic CD3 expression, the T-lineage defining marker, is extremely rare in APL. Atypical morphology and immunophenotype can pose a diagnostic challenge on APL. A diagnosis of MPAL requires exclusion of recurrent cytogenetic abnormities such as t(15;17). Clinical features of APL should prompt immediate testing for the PML-RARA translocation even if atypical immunophenotypic features are present, since early detection of APL may help lower the risk of early death in this disease.
A 59-year-old man presented with oral bleeding. Peripheral blood showed leukocytosis (59.6 × 109/L; 83% blasts). Bone marrow biopsy revealed markedly increased immature cells (panel A; hematoxylin and eosin stain, ×40 objective, original magnification ×400). Aspirate smear showed blasts with round to irregular nuclei, fine chromatin, and small amount of cytoplasm; no obvious Auer rods were identified (panel B; Wright and Giemsa stains, ×100 objective, original magnification ×1000). The blasts were positive for myeloperoxidase (panel C; cytochemical stain, ×100 objective, original magnification ×1000). Flow cytometry showed the blasts (CD34+CD117+HLA-DR−) coexpressed myeloid (myeloperoxidase, CD13, and CD33) and T-lineage (cytoplasmic CD3, CD2, and CD7) markers, suggestive of mixed-phenotype acute leukemia (MPAL), T/myeloid (panels D-E; red cells in panel D are blasts, and blue cells in panel D are T and B cells in the background). However, his D-dimer level was markedly increased (>20 μg/mL fibrinogen equivalent units; normal range 0-0.4), accompanied by a decreased fibrinogen level, concerning for acute promyelocytic leukemia (APL). Fluorescence in situ hybridization confirmed PML/RARA rearrangement (panel F, yellow arrows; original magnification ×1000). Molecular studies revealed an internal tandem duplication and a D835 point mutation of the FLT3 gene. He was diagnosed with microgranular APL.
APL is a hematologic emergency due to its high mortality rate. Cytoplasmic CD3 expression, the T-lineage defining marker, is extremely rare in APL. Atypical morphology and immunophenotype can pose a diagnostic challenge on APL. A diagnosis of MPAL requires exclusion of recurrent cytogenetic abnormities such as t(15;17). Clinical features of APL should prompt immediate testing for the PML-RARA translocation even if atypical immunophenotypic features are present, since early detection of APL may help lower the risk of early death in this disease.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org.