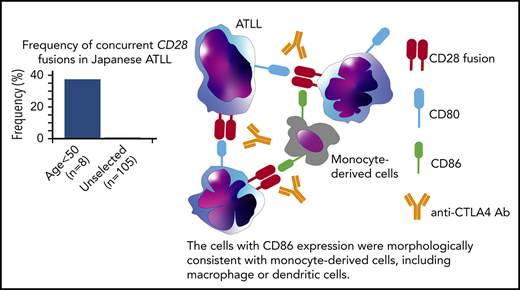
Key Points
Concurrent fusions of CD28 with CTLA4 and ICOS were identified in 3 of 8 Japanese patients <50 years old with ATLL.
Activation of CTLA4-CD28 depends on cell-autonomous and nonautonomous interactions that are targetable with CTLA4 blockade.

Abstract
Adult T-cell leukemia/lymphoma (ATLL) in Japan presents at a median age of 70 years and only 5% of patients are <50 years of age. We conducted RNA and targeted DNA sequencing of 8 ATLLs from Japanese patients <50 years of age and identified 3 (37.5%) with both CTLA4-CD28 and inducible costimulator (ICOS)-CD28 fusions. Mutations of PLCG1, PRKCB, and STAT3, which were frequent in other ATLL-sequencing studies, were not identified. Differential expression analysis identified the negative checkpoint molecule LAG3 as the most downregulated gene among cases with the fusions. Immunohistochemistry demonstrated expression of CD80 and CD86, the ligands for CTLA4 and CD28, on ATLL cells and tumor-associated macrophages, respectively. Expression of CTLA4-CD28 in Ba/F3 cells conferred cytokine-independent growth when cocultured with Raji cells that express CD80 and CD86. Growth was associated with recruitment of the p85 subunit of phosphatidylinositol 3-kinase to CTLA4-CD28 and phosphorylation of AKT and extracellular signal-regulated kinase. A CTLA4-blocking antibody reduced cytokine-independent growth in a dose-dependent manner. Together, these results suggest that young Japanese ATLL cases have a unique biology dependent on cell-nonautonomous interactions that drive CD28 signaling. Assessment for CD28 fusions and treatment with CTLA4 blockade should be considered in younger patients with relapsed/refractory ATLL.
Introduction
Adult T-cell leukemia/lymphoma (ATLL) is a T-cell neoplasm induced by human T-cell leukemia virus type 1. Recent genomic studies have identified recurrent genomic alterations in ATLL that commonly involve T-cell receptor (TCR) signaling and immune escape.1-3 Genomic alterations differ between ATLL subtypes, consistent with specific alterations promoting more aggressive phenotypes.1,4
Outcomes among patients with aggressive subtypes remain poor, indicating a desperate need for therapies that directly target ATLL vulnerabilities.5 In Japan, the median age of ATLL presentation in Japan is >70 years6 of age and only 5% of patients are <50 years old.7,8 In other cancer types, younger age is associated with unique fusions (eg, EML4-ALK in lung adenocarcinoma, ETV6-RUNX1 in B-cell leukemia).9,10 Thus, we hypothesized that ATLL in younger patients will have distinct genetic alterations.
Study design
The studies were approved by the research ethics board at Kurume University. Nucleic acids were extracted from frozen lymph nodes. Transcriptome and DNA-targeted capture sequencing (supplemental Table 1, available on the Blood Web site) were performed and analyzed as previously reported.11 Somatic mutations were called using GeMSTONE and filtered by removing known variants. STAR-Fusion (https://github.com/STAR-Fusion/Star-Fusion) was used for gene fusion detection.
Antibodies for flow cytometry, western blot and immunohistochemistry, and additional details are included in supplemental Methods.
Results and discussion
We performed targeted sequencing of 98 lymphoma-relevant genes11 in ATLL samples from 8 Japanese patients <50 years old (supplemental Tables 2 and 3). The median number of putative mutations was 2 (range, 0-9) (Figure 1A; supplemental Table 4). All mutations except for a single TP53 mutation (case 9060) were present at variant allele frequencies consistent with heterozygosity although we cannot exclude subclonal homozygosity/hemizygosity. The case 9060 TP53 mutation was present at high variant allele frequency consistent with homozygosity. The most frequently mutated genes were CCR4 and CARD11, each in 3 cases (37.5%). Mutations of PLCG1, PRKCB, and STAT3 were frequent in a previous analysis of 370 ATLLs,2 but were not identified in our cases. The previous study2 included 9 ATLL patients <50 years old for mutation analysis with similar frequencies of genomic mutations to our cohort (supplemental Figure 1a).

Genomic alterations in young ATLL cases. (A) Recurrent fusions and mutations identified in ATLL cases. The frequencies of each alteration are summarized in the middle panel comparing the current study with the previous study of 304 cases. (B) Validation of CTLA4-CD28 and ICOS-CD28 fusions by reverse transcription–polymerase chain reaction. (C) Schematic representations of CTLA4-CD28 and ICOS-CD28 fusions. C, C-terminal end; Ex, exon; ICOS, inducible costimulator; N, N-terminal end; SP, signal peptide; TM, transmembrane.
Genomic alterations in young ATLL cases. (A) Recurrent fusions and mutations identified in ATLL cases. The frequencies of each alteration are summarized in the middle panel comparing the current study with the previous study of 304 cases. (B) Validation of CTLA4-CD28 and ICOS-CD28 fusions by reverse transcription–polymerase chain reaction. (C) Schematic representations of CTLA4-CD28 and ICOS-CD28 fusions. C, C-terminal end; Ex, exon; ICOS, inducible costimulator; N, N-terminal end; SP, signal peptide; TM, transmembrane.
Paired-end RNA-sequencing identified in-frame fusions in 7 of 8 cases (87.5%) (supplemental Table 5). Among these, 3 cases (37.5%) harbored both CTLA4-CD28 and inducible costimulator (ICOS)-CD28 fusions. This contrasts with previous reports, in which cases with both fusions were rare among peripheral T-cell lymphoma (0.3%; 1 of 273) and ATLL (1%; 1 of 105).2,12 Cases with and without CD28 fusions had similar survival (supplemental Figure 1b-d), as previously reported in peripheral T-cell lymphomas.12 We confirmed the presence of both fusions in all 3 cases by reverse transcription–polymerase chain reaction and identified genomic breakpoints in 1 case (Figure 1C; supplemental Figure 1e). Isolation of ICOS-CD28 fusion breakpoints is complicated by the extensive length of involved introns.
CTLA4 inhibits TCR signaling while CD28 and ICOS coactivate TCR signaling through interactions with antigen-presenting cells.13 Although both CTLA4 and CD28 share the ligands CD80 and CD86, the affinity to ligands is higher for CTLA4 than for CD28. The structure of the CTLA4-CD28 fusion suggests that the extracellular portion of CTLA4 is expressed on the cell surface where it can interact with CD80 and CD86 to activate signaling through the intracellular CD28 portion (Figure 1C). In contrast to CTLA4-CD28, the ICOS-CD28 fusion links the N-terminal signal peptide of ICOS with the extracellular and intracellular portions of CD28; this should result in CD28 overexpression and ICOS haploinsufficiency (Figure 1C).14
Expression of many kinase oncogenes (eg, BCR-ABL, EML4-ALK) can promote interleukin 3 (IL-3)-independent growth in Ba/F3 cells.15 We transduced Ba/F3 cells with CTLA4-CD28 or a CTLA4-CD28 mutant (mut); the latter harbors 3 amino acid substitutions that abrogate CD28 signaling (supplemental Figure 2a).16 We then cultured these cells in the presence or absence of irradiated Raji cells, which express CD80 and CD86 (Figure 2A; supplemental Figure 2b). Both CTLA4-CD28 and CTLA4-CD28mut were expressed on the surface of Ba/F3 cells after transduction (supplemental Figure 2c).
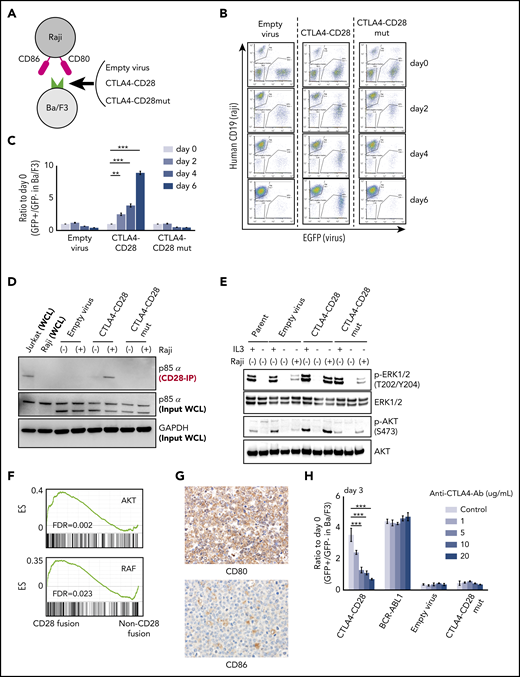
Functional analysis of CTLA4-CD28 fusion. (A) Cartoon depicting an assay in which Ba/F3 cells transduced with empty virus, virus expressing CTLA4-CD28, or virus expressing CTLA4-CD28 with a loss of function (CTLA4-CD28mut) were cocultured with irradiated Raji cells that express CD80 and CD86. (B) Representative results of Ba/F3-Raji assay without mouse IL-3. x-axis indicates enhanced green fluorescent protein (EGFP) as a marker of transduced Ba/F3 cells; y-axis indicates human CD19 expression marking Raji cells. Note that Ba/F3 cells expressing CTLA4-CD28 (GFP+hCD19−) but not Ba/F3 cells transduced with empty virus or CTLA4-CD28mut are present at days 4 and 6. (C) Normalized ratios of EGFP+ fractions in Ba/F3 to those on day 0. The differences were evaluated by 1-way analysis of variance with Bonferroni correction. *P < .05, **P < .01, ***P < .001. Data are presented as means plus or minus standard deviation (SD) from 4 independent experiments, with each performed in triplicate. (D) Western blotting of anti-CD28–immunoprecipitated (IP) samples and whole-cell lysate (WCL) from indicated cell lines. (E) Western blotting of Ba/F3 cells expressing CTLA4-CD28, CTLA4-CD28mut, or empty virus with or without IL3 and Raji cells. (F) GSEA plots of indicated gene sets in the analyzed ATLL cases with or without CD28 fusions. (G) Immunohistochemical features of case 10658 stained for CD80 and CD86. Upper panel shows the image of CD80 stain and lower panel represents that of CD86 stain; original magnification, ×400 for both panels. (H) Normalized ratios of green fluorescent protein–positive (GFP+) Ba/F3 fractions compared with day 0 in the presence of Raji cells and increasing doses of anti-CTLA4 antibody. Data are presented as means plus or minus SD from 4 independent experiments, with each performed in triplicate. The differences were evaluated by 1-way analysis of variance with Bonferroni correction. ***P < .001. Ab, antibody; ES, enrichment score; FDR, false discovery rate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; p-AKT, phosphorylated AKT; p-ERK, phosphorylated ERK.
Functional analysis of CTLA4-CD28 fusion. (A) Cartoon depicting an assay in which Ba/F3 cells transduced with empty virus, virus expressing CTLA4-CD28, or virus expressing CTLA4-CD28 with a loss of function (CTLA4-CD28mut) were cocultured with irradiated Raji cells that express CD80 and CD86. (B) Representative results of Ba/F3-Raji assay without mouse IL-3. x-axis indicates enhanced green fluorescent protein (EGFP) as a marker of transduced Ba/F3 cells; y-axis indicates human CD19 expression marking Raji cells. Note that Ba/F3 cells expressing CTLA4-CD28 (GFP+hCD19−) but not Ba/F3 cells transduced with empty virus or CTLA4-CD28mut are present at days 4 and 6. (C) Normalized ratios of EGFP+ fractions in Ba/F3 to those on day 0. The differences were evaluated by 1-way analysis of variance with Bonferroni correction. *P < .05, **P < .01, ***P < .001. Data are presented as means plus or minus standard deviation (SD) from 4 independent experiments, with each performed in triplicate. (D) Western blotting of anti-CD28–immunoprecipitated (IP) samples and whole-cell lysate (WCL) from indicated cell lines. (E) Western blotting of Ba/F3 cells expressing CTLA4-CD28, CTLA4-CD28mut, or empty virus with or without IL3 and Raji cells. (F) GSEA plots of indicated gene sets in the analyzed ATLL cases with or without CD28 fusions. (G) Immunohistochemical features of case 10658 stained for CD80 and CD86. Upper panel shows the image of CD80 stain and lower panel represents that of CD86 stain; original magnification, ×400 for both panels. (H) Normalized ratios of green fluorescent protein–positive (GFP+) Ba/F3 fractions compared with day 0 in the presence of Raji cells and increasing doses of anti-CTLA4 antibody. Data are presented as means plus or minus SD from 4 independent experiments, with each performed in triplicate. The differences were evaluated by 1-way analysis of variance with Bonferroni correction. ***P < .001. Ab, antibody; ES, enrichment score; FDR, false discovery rate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; p-AKT, phosphorylated AKT; p-ERK, phosphorylated ERK.
In the absence of Raji cells, neither CTLA4-CD28 nor CTLA4-CD28mut expression conferred IL-3–independent growth (supplemental Figure 3a). However, Ba/F3 cells expressing CTLA4-CD28 cultured with Raji cells achieved IL-3 independence (Figure 2B-C). This was not the case for Ba/F3 cells expressing CTLA4-CD28mut (Figure 2B-C), indicating a requirement for functional CTLA4-CD28 signaling. Expression of ICOS-CD28 in Ba/F3 cells also conferred a small but statistically significant growth advantage (supplemental Figure 3b-c), suggesting that ICOS-CD28 provides additional mitogenic signaling.
CD28 is known to recruit the p85α subunit of phosphatidylinositol 3-kinase upon activation and phosphorylation.17 We isolated Ba/F3 cells expressing CTLA4-CD28 in the presence or absence of Raji cells. Coimmunoprecipitation demonstrated binding of p85α with CTLA4-CD28 only with Raji coculture (Figure 2D) associated with phosphorylation of AKT and the MAPK pathway effectors extracellular signal-regulated kinase 1 (ERK1)/ERK2 (Figure 2E).18 These results are consistent with previous results in Jurkat and H9 cells19 but further demonstrate activation of signaling by CTLA4-CD28 in the absence of an endogenous TCR complex.
Gene-set enrichment analysis (GSEA) comparing the 3 ATLL cases with CD28 fusions to the 5 cases that lacked fusions demonstrated enrichment of gene signatures associated with AKT and RAF signaling (Figure 2F). A gene signature that distinguished the cases with and without fusions (supplemental Figure 4a; supplemental Table 6) consisted of 70 genes (log2 fold change > 1; adjusted P < .01). Strikingly, LAG3 was the most significantly downregulated gene among cases with CD28 fusions (log2 fold change, −2.4; P = 10−6) Like CTLA4, LAG3 (CD278) is a transmembrane protein that negatively regulates T-cell proliferation by binding major histocompatibility complex class II.20,21 LAG3 plays an important role in the function of regulatory T cells, which are the immunophenotypic counterparts of ATLL.22,23 GSEA also identified enrichment of interferon response and other T-cell function–related signatures in cases with CD28 fusions (supplemental Table 7).
Cases with CTLA4-CD28 and ICOS-CD28 fusions had higher expression of CD80 transcript (supplemental Figure 4b). By immunohistochemistry, ATLL cells with CTLA4-CD28 and ICOS-CD28 fusions expressed surface CD80, and macrophages in the tumor microenvironment expressed CD86 (Figure 2G). Thus, both intracellular and intercellular interactions could drive CTLA4-CD28 and ICOS-CD28 signaling, although more extensive analyses will be needed to determine the frequency of CD80/CD86 expression in the ATLL microenvironment. In addition, further studies are clearly needed to address the molecular epidemiology of CD28 fusions in larger cohorts and other geographic regions. The availability of next-generation sequencing platforms that use paraffin-embedded biopsies and can identify CD28 fusions makes it possible to broadly assess for these alterations in patients with relapsed/refractory ATLL.24 However, single-cell RNA sequencing at high-depth will likely be required to define the frequencies at which individual ATLL cells harbor ICOS-CD28, CTLA4-CD28, or both.
Finally, a previous case report described a patient with Sezary syndrome and CTLA4-CD28 fusion who had a deep but transient response to the anti-CTLA4 antibody ipilimumab.25 This study did not clarify the extent to which response resulted from blocking of cell-autonomous CTLA4-CD28 signaling vs activation of a cell nonautonomous immune response. Treatment of Ba/F3 cells expressing CTLA4-CD28 and cultured in the presence of Raji cells with a CTLA4-blocking antibody suppressed proliferation in a dose-dependent fashion (Figure 2H). The same antibody had no effect on Ba/F3 cells expressing BCR-ABL cultured in the presence of Raji cells, confirming that the effect is specific to CTLA4-CD28. Based on these studies, agents that block CTLA4 or target downstream signaling26,27 merit testing in cases of ATLL with CD28 fusions.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE143986).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
The authors thank Hiroaki Miyoshi for generous technical assistance in these studies.
N.Y. was supported by the Japanese Society for the Promotion of Science. This work was supported by Leukemia & Lymphoma Society Specialized Center of Research #7011-16 and National Institutes of Health, National Cancer Institute grant R35 CA231958 (D.M.W.) and Japanese Society for the Promotion of Science KAKENHI grant number JP19K16574 (N.Y.).
Authorship
Contribution: N.Y. designed, executed, and analyzed the experiments; K.S., N.D., C.T., N.A.C., X.B., F.A., M.T., K.O., and A.Y. assisted in performing the experiments; K.E.S. assisted with analysis; S.Y.N. and D.M.W. designed and analyzed the experiments; and N.Y. and D.M.W. cowrote the manuscript.
Conflict-of-interest disclosure: D.M.W. receives research support from Verastem; AstraZeneca, Aileron, Abbvie, Daiichi Sankyo, Verastem and Surface Oncology, and consulting or Scientific Advisory Board Membership from Travera, Mundipharma EDO, Bantam, Ajax, Genentech/Roche, Dragonfly, Prescient, Ossium, and Myeloid Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: David M. Weinstock, Dana-Farber Cancer Institute, 450 Brookline Ave, Dana 510B, Boston, MA 02215; e-mail: dweinstock@partners.org; or Noriaki Yoshida, Radiation Effects Research Foundation, 5-2 Hijiyama Park, Minami-ku, Hiroshima, 7320815 Japan; e-mail: noriaki3@rerf.or.jp.