Key Points
The mechanisms of anti-GPIbα antibody-induced thrombocytopenia and thrombopoietin response differ in relation to antibody dose.
Megakaryocytes targeted by anti-GPIbα antibody generate platelets with reduced surface GPIbα expression.

Abstract
Immune thrombocytopenia (ITP) is an acquired bleeding disorder characterized by antibody-mediated platelet destruction. Different mechanisms have been suggested to explain accelerated platelet clearance and impaired thrombopoiesis, but the pathophysiology of ITP has yet to be fully delineated. In this study, we tested 2 mouse models of immune-mediated thrombocytopenia using the rat anti-mouse GPIbα monoclonal antibody 5A7, generated in our laboratory. After a single IV administration of high-dose (2 mg/kg) 5A7, opsonized platelets were rapidly cleared from the circulation into the spleen and liver; this was associated with rapid upregulation of thrombopoietin (TPO) messenger RNA. In contrast, subcutaneous administration of low-dose 5A7 (0.08-0.16 mg/kg) every 3 days gradually lowered the platelet count; in this case, opsonized platelets were observed only in the spleen, and TPO levels remained unaltered. Interestingly, in both models, the 5A7 antibody was found on the surface of, as well as internalized to, bone marrow megakaryocytes. Consequently, platelets generated in the chronic phase of repeated subcutaneous 5A7 administration model showed reduced GPIbα membrane expression on their surface. Our findings indicate that evaluation of platelet surface GPIbα relative to platelet size may be a useful marker to support the diagnosis of anti-GPIbα antibody–induced ITP.
Introduction
Human and animal studies indicate that the pathology and clinical course of immune thrombocytopenia (ITP) caused by autoantibodies against platelet glycoproteins varies, depending on the target antigen.1,2 Studies of mouse models show differences in the response to treatments depending on the pathogenic autoantibodies.3 Clinical studies report that resistance to IV immunoglobulin (IVIG) treatment is more frequent when ITP is caused by anti-GPIb/IX compared with anti-GPIIb-IIIa (integrin αIIbβ3) autoantibodies, possibly because of different platelet clearance mechanisms.4,5 Aging platelets that become desialylated are removed from the circulation in the liver via the Ashwell-Morell receptor (AMR) in a process that regulates thrombopoietin (TPO) production by hepatocytes.6 Likewise, platelets opsonized with anti-GPIbα antibodies are activated and desialylated after neuraminidase-1 translocation to the membrane, resulting in Fc-independent hepatic clearance via the AMR.7 In addition, GPIbα, particularly the amino-terminal domain, has been associated with the production of hepatic TPO, and anti-GPIbα antibodies can impair platelet-mediated TPO expression by cultured hepatocytes.8 The significance of clearance processes in vivo, however, is still unclear.7,9,10 We have used 2 different methods to induce thrombocytopenia in mice with anti-GPIbα antibodies and found distinct organ-specific consequences on platelet clearance and TPO production, as well as altered thrombocytopoiesis by megakaryocytes (MKs) targeted with anti-GPIbα.
Material and methods
Reagents
Anti-GPIbα R300 and DyLight 649–labeled Xia.G5 were from Emfret Analytics (Eibelstadt, Germany). PE-Cy7–labeled goat anti-rat IgG and Brilliant Violet 421–labeled anti-αIIb (MWReg30) were from BioLegend (San Diego, CA). AlexaFluor 555-labeled goat anti-rabbit IgG and AlexaFluor 488–labeled goat anti-rat IgG were from Invitrogen (Carlsbad, CA). Anti-F4/80 antibody (CI: A3-1) was from Absolute Antibody (Boston, MA). Rabbit anti-ASGPR1 antibody (50083-R114) was from Sino Biological (Wayne, PA). IVIG (Gammagard, 10%) was from Baxalta US Inc (Lexington, MA).
Animal study
Animal experiments were performed according to a protocol approved by The Scripps Research Institutional Animal Care and Usage Committee.
Messenger RNA quantification
Total RNA was extracted from homogenized liver by using Trizol reagent (Invitrogen) and purified using Monarch Total RNA Miniprep Kit (New England BioLabs, Ipswich, MA). Complementary DNAs were synthesized with SuperScript III (Invitrogen). The assay IDs (Integrated DNA Technologies, Skokie, IL) for primers and probes were as follows: Thpo (Mm.PT.58.17230736), Il1a (Mm.PT.58.32778767), and B2M (Mm.PT.39a.22214835). Relative gene expressions were calculated according to the comparative Ct method using B2M as an internal control.
Platelet analysis
Complete blood count was obtained with the Procyte Dx (IDEXX Laboratories, Westbrook, ME).
Fluorescence-activated cell sorting analysis
Blood samples were fixed with 2% paraformaldehyde and stained with PE-Cy7–labeled anti-rat IgG to detect surface-bound 5A7. After removal of the unbound anti-rat IgG, GPIbα and GPIIb (αIIb) were stained with Xia.G5 (Emfret) and MWReg30 (BioLegend), respectively. Samples were analyzed on a Novocyte flow cytometer (ACEA Biosciences). The results were analyzed with FlowJo software.
Histology
Cryosections were prepared as previously described,11,12 fixed, stained, and visualized in a fluorescence microscope (BZ-X700; Keyence, Woodcliff Lake, NJ) or confocal microscope (LSM 880; Carl Zeiss, Thornwood, NY). In brief, the harvested organs were snap frozen after perfusion with phosphate-buffered saline and cryosectioned using the modified Kawamoto’s method. After fixation with 4% paraformaldehyde, samples were blocked and permeabilized using 1% Triton X-100 PHEM buffer containing 5% normal goat serum and FcR blocker (Innovex Bioscience, Richmond, CA). F4/80 was stained with rabbit anti-F4/80 (CI: A3-1) and visualized with AlexaFluor 555–labeled goat anti-rabbit IgG. Injected 5A7 was detected with AlexaFluor 488–labeled goat anti-rat IgG. After absorbing anti-rat secondary antibody and extensive washing, the samples were sequentially stained with biotinylated rat anti-αIIb antibody (MWReg30) followed by streptavidin-AlexaFluor 647 (Biolegend). The nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI).
Results
Generation and analysis of acute platelet-depletion model by IV administration of anti-GPIbα antibody
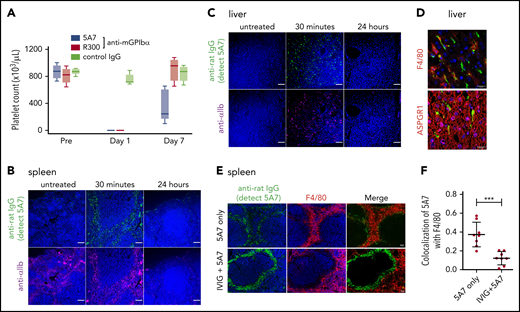
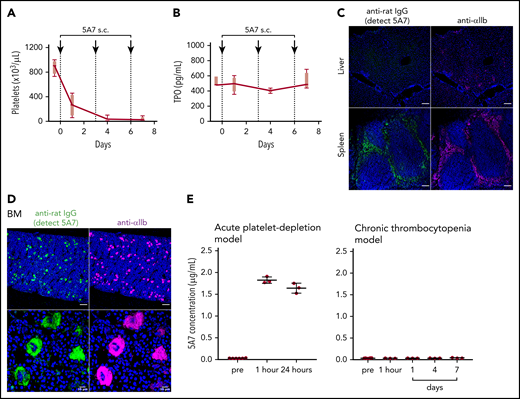
For these studies, we used the anti-mouse GPIbα rat monoclonal antibody (mAb) 5A7, generated in our laboratory, which causes platelet depletion in vivo13 and inhibits platelet adhesion to von Willebrand factor (VWF) under flow (supplemental Figure 1, available on the Blood Web site). In a first set of experiments, we injected 2 mg/kg of 5A7 intravenously to achieve rapid anti-GPIbα antibody-induced thrombocytopenia. The platelet count was below the detection limit of an automated blood cell counter at day 1 and still lower than in the controls at day 7 (Figure 1A). By immunofluorescence analysis with an anti-rat IgG antibody, opsonized platelets with bound 5A7 were found in the spleen (Figure 1B) and liver (Figure 1C) 30 minutes after antibody injection, and were markedly decreased at 24 hours. Platelets observed in the liver tended to form aggregates, some associated with or inside F4/80+ cells, whereas others were detected adjacent to AMR+ cells (Figure 1D).
Generation and analysis of an acute platelet-depletion model induced by single high-dose injection of an anti-GPIbα antibody. (A) An anti-GPIbα antibody (5A7 or R300, 2 mg/kg) was administered to C57BL/6J wild-type (WT) mice by retro-orbital injection, and platelet counts were monitored. (B-C) Platelet distribution was visualized by wide-field immunofluorescence analysis of cryosections prepared from spleen (B) and liver (C) harvested from untreated mice or from treated mice 30 minutes or 24 hours after antibody administration (BZ-X700 microscope). Bars represent 100 µm. Tissues were stained with DAPI (blue), anti-rat IgG (green; detecting administered 5A7 antibody), and anti-αIIb antibody (purple). (D) Immunofluorescence staining of cryosectioned liver. Opsonized platelets were stained with anti-rat IgG (green) and macrophages were stained with anti-F4/80 antibody (red). Hepatocytes were stained with anti-ASGPR1 antibody (red; bottom). Images were obtained by confocal microscopy (LSM 880 microscope). Bars represent 20 µm. (E) IVIG was administered via intraperitoneal injection (2 g/kg) 24 hours before 5A7 IV injection (0.6 mg/kg). Spleens harvested 20 minutes after 5A7 administration were cryosectioned and immunostained with anti-rat IgG (green; detecting administered 5A7 antibody) and anti-F4/80 antibody (red), along with costaining with DAPI (blue). (F) Images were binarized, and colocalization of opsonized platelets (stained by anti-rat IgG) with F4/80 was calculated as a ratio by quantifying anti-rat IgG and F4/80 costained area divided by total of anti-rat IgG–stained area, by using Fiji image-analysis software. ***P < .001 by unpaired Student t test.
Generation and analysis of an acute platelet-depletion model induced by single high-dose injection of an anti-GPIbα antibody. (A) An anti-GPIbα antibody (5A7 or R300, 2 mg/kg) was administered to C57BL/6J wild-type (WT) mice by retro-orbital injection, and platelet counts were monitored. (B-C) Platelet distribution was visualized by wide-field immunofluorescence analysis of cryosections prepared from spleen (B) and liver (C) harvested from untreated mice or from treated mice 30 minutes or 24 hours after antibody administration (BZ-X700 microscope). Bars represent 100 µm. Tissues were stained with DAPI (blue), anti-rat IgG (green; detecting administered 5A7 antibody), and anti-αIIb antibody (purple). (D) Immunofluorescence staining of cryosectioned liver. Opsonized platelets were stained with anti-rat IgG (green) and macrophages were stained with anti-F4/80 antibody (red). Hepatocytes were stained with anti-ASGPR1 antibody (red; bottom). Images were obtained by confocal microscopy (LSM 880 microscope). Bars represent 20 µm. (E) IVIG was administered via intraperitoneal injection (2 g/kg) 24 hours before 5A7 IV injection (0.6 mg/kg). Spleens harvested 20 minutes after 5A7 administration were cryosectioned and immunostained with anti-rat IgG (green; detecting administered 5A7 antibody) and anti-F4/80 antibody (red), along with costaining with DAPI (blue). (F) Images were binarized, and colocalization of opsonized platelets (stained by anti-rat IgG) with F4/80 was calculated as a ratio by quantifying anti-rat IgG and F4/80 costained area divided by total of anti-rat IgG–stained area, by using Fiji image-analysis software. ***P < .001 by unpaired Student t test.
To determine the potential of 5A7 to induce platelet aggregation, mouse whole-blood samples were treated with 5A7 ex vivo and analyzed by fluorescence-activated cell sorting (FACS). Platelet agglutination was observed with 5A7 treatment, and the percentage was significantly higher than in those treated with rat mAb against integrin αIIb (supplemental Figure 2A-B). IgG is bivalent, and therefore, 5A7 antibody binding can physically cross-link platelets. In addition, 5A7 binds to the N-terminal VWF binding region of mGPIbα (supplemental Figure 1). Thus, the distance from the plasma membrane may enable efficient cross-linking of platelets to one another compared with anti-integrin αIIb antibody, even at static conditions.14,15 It has been reported that antibodies against the ligand-binding domain of GPIbα causes shear-dependent platelet activation and desialylation.16 To examine whether in vivo administered 5A7 binds to platelets and induces desialylation, different doses of 5A7 (0.2 and 0.6 mg/mL) were administered so that a small number of platelets remained in circulation to be analyzed by FACS. The results show a dose-dependent increase in RCA-1 binding, supporting our hypothesis that 5A7 binding causes platelet desialylation and AMR binding in the liver (supplemental Figure 2C). To confirm colocalization of opsonized platelets with AMR+ cells, asialofetuin was administered 20 minutes before 5A7 IV injection to block AMR. Immunofluorescence staining and quantitative analysis showed significantly reduced colocalization of opsonized platelets with AMR when treated with asialofetuin (supplemental Figure 3A-B). FACS analysis of blood samples showed the presence of platelet aggregates induced by 5A7 injection (supplemental Figure 3C). Treatment with asialofetuin did not make a significant difference in the percentage of platelet aggregates induced by 5A7 (supplemental Figure 3D), suggesting that platelet aggregates stuck in liver microvasculature may not recirculate, but instead are cleared by Kupffer cells. Administration of asialofetuin blocked AMR, resulting in the increase in the percentage of desialylated platelets in circulation, although the difference was not statistically significant (supplemental Figure 3E-F). These results confirm IV injection of 5A7 induces platelet aggregation and desialylation, leading to AMR binding and hepatic sequestration.
Platelets in the spleen were accumulated in the region where F4/80+ cells were enriched (Figure 1E), suggesting that these platelets were captured by F4/80+ macrophages and/or dendritic cells via Fc receptors. To confirm this result, mice were treated with IVIG before 5A7 administration. Localization of opsonized platelets was altered by pretreatment with IVIG, and colocalization of opsonized platelets with F4/80 was significantly reduced (Figure 1E-F). IVIG treatment significantly increased the number of platelets remaining in circulation 30 minutes after 5A7 injection (24.0 × 103/μL ± 7.0 × 103/μL; n = 4) compared with mice treated with 5A7 only (1.0 × 103/μL ± 1.4 × 103/μL; n = 4). Although the difference was significant (P = .0286, by Student t test), the increased platelet count corresponds to only 3% of normal platelet number, indicating a minimum overall therapeutic effect of IVIG on thrombocytopenia in this 5A7 IV injection model.
Hepatic TPO expression is upregulated in the anti-GPIbα antibody–induced acute platelet-depletion model
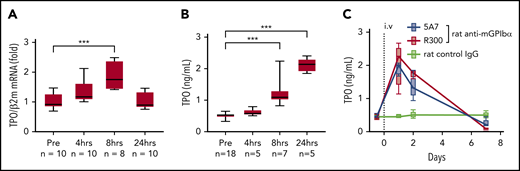
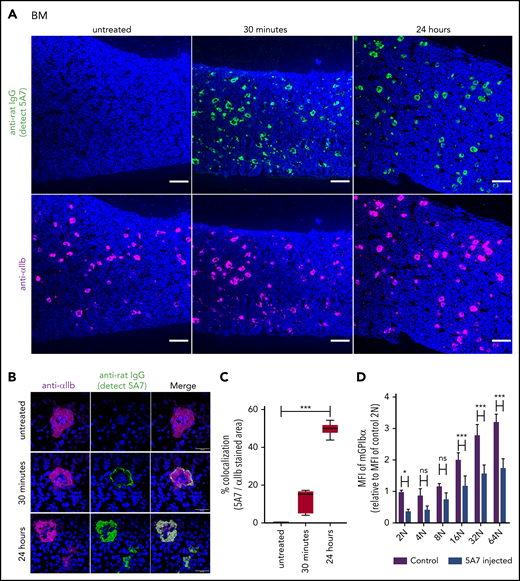
Based on the results of fluorescence immunostaining, it was speculated that high-dose injection of the antibody induces platelet activation, aggregation, and desialylation, which may stimulate TPO production by signaling through hepatocyte AMR. To test our hypothesis, TPO messenger RNA (mRNA) expression was measured by real-time polymerase chain reaction (PCR) using liver tissue harvested at different time points after 5A7 injection. As expected, TPO mRNA level was increased at 4 hours, peaked at 8 hours, and returned to normal by 24 hours (Figure 2A). Plasma TPO levels were significantly higher at 8 and 24 hours after injection (Figure 2B). To confirm these findings, which are in contrast with a previous report,8 we repeated the study with a different rat anti-mouse GPIbα mAb, R300, obtained commercially.17 The decrease in platelets caused by 5A7 and R300 was similar at day 1, and full recovery was observed at day 7 (Figure 1A). TPO plasma levels increased similarly, peaking at day 1 and returning to baseline by day 7 (Figure 2C). Thus, stimulation of hepatic TPO production was confirmed with 2 different anti-GPIbα antibodies. Interestingly, 30 minutes after injection, 5A7 was not only associated with circulating platelets, but also MKs in the bone marrow (BM; Figure 3A). At 24 hours after injection, 5A7 was internalized within MKs (Figure 3B). Quantitative analysis showed enhanced colocalization of 5A7 with integrin αIIb, which is known to be present, not only on the surface of MKs, but also inside, at 24 hours after 5A7 administration (Figure 3C). We evaluated surface expression of GPIbα in the BM MKs 24 hours after 5A7 injection. As expected, expression of mGPIbα was reduced by half vs controls (Figure 3D). This result indicates that in vivo–administered 5A7 was bound to mGPIbα on MK and was internalized, leading to reduced mGPIbα expression on the MK surface. Total number of MKs and ploidy distribution were unchanged (supplemental Figure 4). To test 5A7 binding to mGPIbα on MKs and internalization in vitro, CD41+ cells were isolated from C57BL/6J wild-type mouse BM and transduced with LIM-homeobox 2 (Lhx2) retrovirus to facilitate in vitro expansion. CD41+ Lhx2 hematopoietic progenitors were then cultured in the presence of TPO, stem cell factor, and interleukin-6 (IL-6), to differentiate mature MKs.13,18 Cells were transferred to serum-free medium and AlexaFluor 647–labeled 5A7 was added into in vitro MK culture. The binding of 5A7 to MKs was observed after 15 minutes, and 5A7 internalization gradually increased in the subsequent 24 hours (supplemental Figure 5).
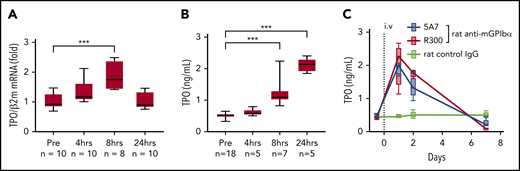
Upregulation of TPO in the acute platelet-depletion model. (A) Liver TPO mRNA was quantified by real-time quantitative PCR before and at the indicated time points after IV 5A7 injection. (B-C) Plasma TPO was measured by enzyme-linked immunosorbent assay before (B) and after (C) antibody injection, as indicated. ***P < .001 by 1-way analysis of variance, with Dunn’s multiple-comparison test.
Upregulation of TPO in the acute platelet-depletion model. (A) Liver TPO mRNA was quantified by real-time quantitative PCR before and at the indicated time points after IV 5A7 injection. (B-C) Plasma TPO was measured by enzyme-linked immunosorbent assay before (B) and after (C) antibody injection, as indicated. ***P < .001 by 1-way analysis of variance, with Dunn’s multiple-comparison test.
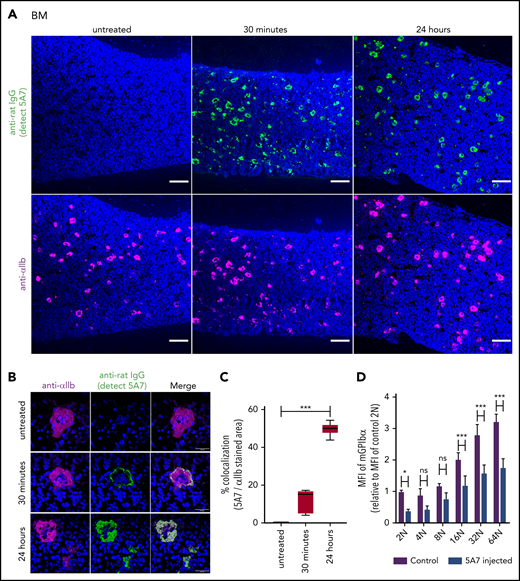
Analysis of BM MKs in the acute platelet-depletion model. (A-C) BM sections harvested at the indicated time points after IV 5A7 injection were stained with DAPI (blue), anti-rat IgG (green; detecting administered 5A7 antibody), and anti-αIIb antibody (purple) and visualized by BZ-X700 microscope (bars represent 100 µm) (A) or confocal microscopy (LSM 880 microscope; bars represent 10 µm) (B). (C) Images were binarized, and colocalization of 5A7 with integrin αIIb was calculated as a percentage by quantifying the anti-rat IgG–stained area and dividing by the anti-integrin αIIb antibody–stained area, by using Fiji image-analysis software. ***P < .001 by one-way analysis of variance with Dunn’s multiple-comparison test. (D) BM cells were harvested 24 hours after IV 5A7 injection and analyzed by FACS for GPIbα expression by staining with monoclonal anti-GPIbα antibody (Xia.G5) which does not compete with 5A7 for binding to GPIbα. *P < .05, ***P < .001 by 2-way analysis of variance with Sidak’s multiple-comparison test.
Analysis of BM MKs in the acute platelet-depletion model. (A-C) BM sections harvested at the indicated time points after IV 5A7 injection were stained with DAPI (blue), anti-rat IgG (green; detecting administered 5A7 antibody), and anti-αIIb antibody (purple) and visualized by BZ-X700 microscope (bars represent 100 µm) (A) or confocal microscopy (LSM 880 microscope; bars represent 10 µm) (B). (C) Images were binarized, and colocalization of 5A7 with integrin αIIb was calculated as a percentage by quantifying the anti-rat IgG–stained area and dividing by the anti-integrin αIIb antibody–stained area, by using Fiji image-analysis software. ***P < .001 by one-way analysis of variance with Dunn’s multiple-comparison test. (D) BM cells were harvested 24 hours after IV 5A7 injection and analyzed by FACS for GPIbα expression by staining with monoclonal anti-GPIbα antibody (Xia.G5) which does not compete with 5A7 for binding to GPIbα. *P < .05, ***P < .001 by 2-way analysis of variance with Sidak’s multiple-comparison test.
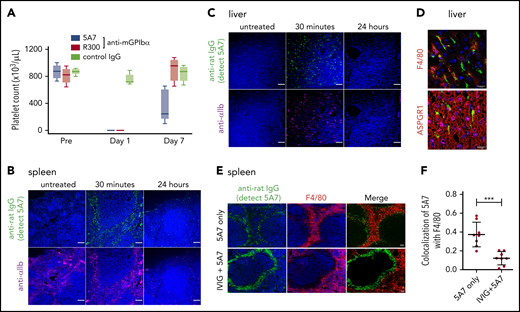
Repeated subcutaneous 5A7 injection generates a chronic thrombocytopenia model
To evaluate the effect of variable anti-GPIbα antibody dose and mode of administration on induced thrombocytopenia, we injected 0.08 mg/kg 5A7 subcutaneously every 3 days for 3 doses. The platelet count dropped by ∼70% at 24 hours after initiating treatment and by ∼95% at day 4, then remained stable for 3 additional days (Figure 4A). Notably, plasma TPO levels were essentially unchanged at all postinjection time points tested (Figure 4B). Moreover, platelets with bound 5A7 were found predominantly in the spleen and not in the liver (Figure 4C), whereas 5A7 binding to BM MKs, both on the surface and internalized, was comparable, regardless of dose and mode of administration (compare Figures 4D and 3B). These findings indicate that the mechanism of platelet clearance in these models differs and that a higher level of anti-GPIbα antibody may be necessary to induce platelet activation/desialylation, leading to hepatic clearance, as compared with splenic sequestration. In support of this hypothesis, plasma concentration of in vivo–administered 5A7 was measured as 1.83 ± 0.07 μg/mL (n = 3) in the acute platelet-depletion model at 1 hour, and the level was maintained at 24 hours after injection (Figure 4E, left). In contrast, 5A7 in the plasma of low-dose subcutaneous injection model was undetectable during the observed period (Figure 4E, right). 5A7 doses used in the subcutaneous injection model (0.08 mg/kg) was lower than the dose used in the acute platelet-depletion model (2 mg/kg). Thus, we sought to determine whether the difference in the hepatic clearance and TPO upregulation depended on the mode of administration or the injected dose. Mice were intravenously injected with different doses of 5A7, and plasma TPO levels were determined at 24 hours after injection. The plasma TPO level was upregulated in a dose-dependent manner, and no significant increase was observed with the lowest dose of 5A7 (0.16 mg/kg; supplemental Figure 6A). Consistent with this result, 5A7 administered in vivo could not be detected at this dose (0.16 mg/kg; supplemental Figure 6B). These results suggest that there is a threshold in the plasma 5A7 concentration required to induce AMR signaling and hepatic clearance and that IV injection is more potent to achieve higher plasma concentrations than subcutaneous injection because of the difference in the mode of administration and absorption.
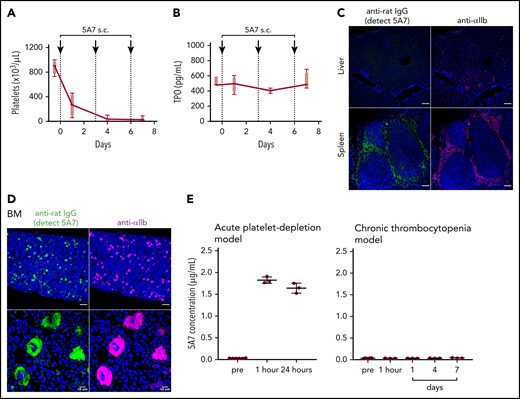
Thrombocytopenia, induced by repeated subcutaneous administration of anti-GPIbα antibody 5A7. (A) The antibody 5A7 (0.08 mg/kg) was injected subcutaneously (s.c.) every 3 days. Blood platelet counts were measured before and at the indicated time points after antibody injection. (B) Plasma TPO level was measured by enzyme-linked immunosorbent assay. (C) Immunostaining analysis of organs harvested on day 8 performed as described for the corresponding images in Figure 1. Bars represent 100 µm. (D) Immunostained BM sections were visualized by fluorescence microscopy (top) and confocal microscopy (bottom). Bars represent 100 μm (upper panels) and 10 μm (lower panels). (E) Plasma samples collected from mice treated with single dose (2 mg/kg) of IV 5A7 injection (left), and 3 doses of subcutaneous injection (0.08 mg/kg each) were analyzed by enzyme-linked immunosorbent assay to measure plasma concentration of administered 5A7.
Thrombocytopenia, induced by repeated subcutaneous administration of anti-GPIbα antibody 5A7. (A) The antibody 5A7 (0.08 mg/kg) was injected subcutaneously (s.c.) every 3 days. Blood platelet counts were measured before and at the indicated time points after antibody injection. (B) Plasma TPO level was measured by enzyme-linked immunosorbent assay. (C) Immunostaining analysis of organs harvested on day 8 performed as described for the corresponding images in Figure 1. Bars represent 100 µm. (D) Immunostained BM sections were visualized by fluorescence microscopy (top) and confocal microscopy (bottom). Bars represent 100 μm (upper panels) and 10 μm (lower panels). (E) Plasma samples collected from mice treated with single dose (2 mg/kg) of IV 5A7 injection (left), and 3 doses of subcutaneous injection (0.08 mg/kg each) were analyzed by enzyme-linked immunosorbent assay to measure plasma concentration of administered 5A7.
Anti-GPIbα antibody does not affect megakaryopoiesis in the BM up to day 8
Another intriguing finding was that anti-GPIbα antibody (5A7) binds, not only to platelets in circulation, but also to MKs in the BM, and becomes internalized. A previous study using the plasma of patients with ITP caused by anti-GPIbα antibodies showed an inhibitory effect on megakaryopoiesis.19 Thus, we set out to examine how anti-GPIbα antibody accumulation in BM MKs influences megakaryopoiesis. To avoid the influence of TPO upregulation, which is not usually observed in patients with ITP, we used the low-dose 5A7 subcutaneous administration as a model to better represent patients with ITP. To study the consequences of MK targeting by anti-GPIbα antibody, BM MKs were analyzed on day 8. Total MK number and MK ploidy distribution were indistinguishable from control BM (supplemental Figure 7). These results show that anti-GPIbα antibody 5A7 binds to and becomes internalized in MKs, but does not affect megakaryopoiesis in the BM up to day 8.
It has been reported that administration of anti-GPIbα antibody causes MK rupture in mice, leading to rapid platelet release.20 The process of platelet production by MK rupture, mediated by IL-1α, has been shown to be quite different from what we call “proplateletlike” platelet production. To examine whether 5A7 dose and administration methods have different effects on IL-1α expression, total BM cells isolated from the acute platelet-depletion mouse model, chronic thrombocytopenia mouse model, as well as untreated control mice (n = 3 in each group) were analyzed. IL-1α mRNA expression in the BM was upregulated in the acute platelet-depletion model, although the difference was not significant (supplemental Figure 8). Upregulation of IL-1α mRNA was not observed with the chronic thrombocytopenia mouse model, indicating that MKs in the BM are under the influence of IL-1α in the acute phase, but not in the chronic phase, of thrombocytopenia.
Platelets in a chronic thrombocytopenia model present reduced GPIbα expression by shedding and internalization
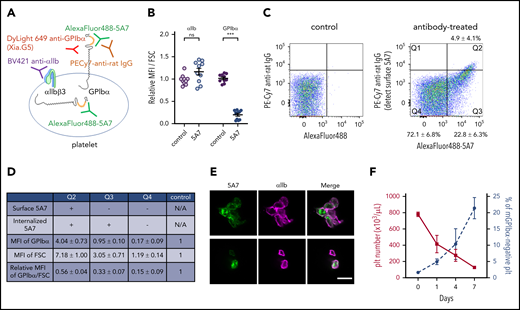
Next, we analyzed platelets produced from 5A7-targeted MKs. Subcutaneous injection of AlexaFluor 488–labeled 5A7 (0.16 mg/kg to compensate for decreased platelet depletion activity after labeling) allowed platelet visualization by FACS in blood samples collected 8 days after the first antibody injection. With this approach, a positive 5A7 signal indicated both surface-bound and internalized antibody. To differentiate the two, surface-bound 5A7 was detected with PE-Cy7 labeled anti-rat IgG. Platelet GPIbα was quantified with an antibody Xia.G5, which does not compete with 5A7 for binding to mouse GPIbα (Figure 5A). Platelets were, on average, 1.6 times larger, as measured by increased forward scatter (FSC), than those of control mice that were not injected with 5A7. Thus, for quantification, mean fluorescence intensity (MFI) values were divided by the FSC and presented as a ratio relative to the control. The calculated GPIbα surface expression was significantly lower in mice with anti-GPIbα–induced thrombocytopenia than in controls, whereas integrin αIIb was comparable (Figure 5B). Based on the presence/absence and subcellular localization of in vivo–administered 5A7, 3 platelet populations were identified (Figure 5C). Only 4.9% ± 4.1% (cytometry plot quadrant [Q]2) had surface-bound antibody 5A7, as positive for both AlexaFluor 488-5A7 signal and also detected by PE-Cy7 labeled anti-rat IgG. Platelets in the Q3 fraction (22.8% ± 6.3%) had internalized 5A7 as evidenced by the presence of AlexaFluor 488–labeled 5A7 which are not stained by PE-Cy7 labeled anti-rat IgG. A majority (72.1% ± 6.8%) had no 5A7, either on the surface or within the cytoplasm (Q4). Of note, most of the Q2 platelets with membrane-bound 5A7 formed aggregates, as shown by an FSC 7.2 times greater than the control and confocal microscopy (Figure 5D-E, top). Single platelets were present in Q3 and Q4 (Figure 5E, bottom); the former was larger than the latter (FSC 3.1 vs 1.2 times control). Expression of GPIbα divided by the FSC in each platelet population is shown in the bottom row; Q4 showed markedly lower GPIbα on the membrane surface (0.2 times control; Figure 5D). To further characterize newly generated platelets in our chronic thrombocytopenia model, the percentage of platelets that lost surface GPIbα expression, by shedding or internalization, was evaluated. The percentage of platelets that lost surface GPIbα expression gradually increased during the observation period of 7 days, suggesting that these GPIbα-negative platelets are, at least partially, produced from MKs that had been targeted by 5A7 in the BM (Figure 5F).
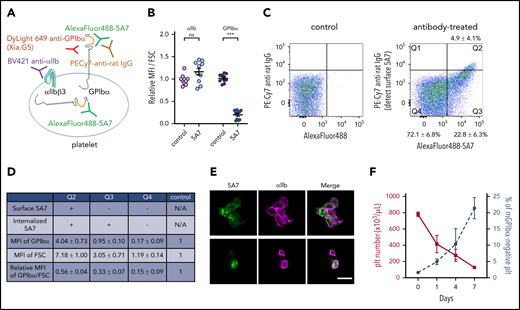
Analysis of platelets in thrombocytopenia mice induced by subcutaneous 5A7 injection. (A) Schematic diagram of platelet staining for FACS analysis. In this experiment, AlexaFluor 488–labeled 5A7 was administered in vivo so it could be detected regardless of subcellular localization. The platelet population was gated using a Brilliant Violet (BV) 421–labeled anti-αIIb antibody. PE-Cy7 labeled anti-rat IgG was used to detect 5A7 bound to GPIbα on the platelet surface. DyLight649 anti-GPIbα antibody (Xia.G5) was used to stain GPIbα on the platelet surfaces. (B) MFI of platelet GPIbα and αIIb corrected for platelet size (FSC) and expressed relative to normal control. Total results of 2 independent experiments are shown (n = 10 in each group); ***P < .001 determined by Student t test. NS, not significant. (C) Platelet subpopulations identified by presence/absence of in vivo–administered AlexaFluor 488–labeled 5A7 and their subcellular location. Positive signals on the abscissa indicate the presence of 5A7 either inside or on the surface of platelets. Positive signals on the ordinate indicate the presence of 5A7 on the platelet surface. The criteria of the quadrant were determined by a control blood sample collected from an untreated mouse stained with PE-Cy7 labeled anti-rat IgG (left). Mean percentage and standard deviations were calculated for the 10 mice tested. (D) GPIbα expression on platelet surface was determined by staining with DyLight 649 anti-GPIbα antibody (Xia.G5) which does not compete with 5A7 for GPIbα binding. The MFI of each population is indicated as relative to normal controls. MFI of GPIbα corrected by platelet size (FSC) is shown in the bottom row of the table. Results are obtained from 10 mice. (E) Confocal analysis of platelets in Q2 (top) and Q3/Q4 (bottom) subpopulations. Bar represents 5 µm. (F) During the course of chronic thrombocytopenia induced by 5A7 subcutaneous administration every 3 days, the percentage of platelets negative for surface GPIbα expression was monitored by FACS.
Analysis of platelets in thrombocytopenia mice induced by subcutaneous 5A7 injection. (A) Schematic diagram of platelet staining for FACS analysis. In this experiment, AlexaFluor 488–labeled 5A7 was administered in vivo so it could be detected regardless of subcellular localization. The platelet population was gated using a Brilliant Violet (BV) 421–labeled anti-αIIb antibody. PE-Cy7 labeled anti-rat IgG was used to detect 5A7 bound to GPIbα on the platelet surface. DyLight649 anti-GPIbα antibody (Xia.G5) was used to stain GPIbα on the platelet surfaces. (B) MFI of platelet GPIbα and αIIb corrected for platelet size (FSC) and expressed relative to normal control. Total results of 2 independent experiments are shown (n = 10 in each group); ***P < .001 determined by Student t test. NS, not significant. (C) Platelet subpopulations identified by presence/absence of in vivo–administered AlexaFluor 488–labeled 5A7 and their subcellular location. Positive signals on the abscissa indicate the presence of 5A7 either inside or on the surface of platelets. Positive signals on the ordinate indicate the presence of 5A7 on the platelet surface. The criteria of the quadrant were determined by a control blood sample collected from an untreated mouse stained with PE-Cy7 labeled anti-rat IgG (left). Mean percentage and standard deviations were calculated for the 10 mice tested. (D) GPIbα expression on platelet surface was determined by staining with DyLight 649 anti-GPIbα antibody (Xia.G5) which does not compete with 5A7 for GPIbα binding. The MFI of each population is indicated as relative to normal controls. MFI of GPIbα corrected by platelet size (FSC) is shown in the bottom row of the table. Results are obtained from 10 mice. (E) Confocal analysis of platelets in Q2 (top) and Q3/Q4 (bottom) subpopulations. Bar represents 5 µm. (F) During the course of chronic thrombocytopenia induced by 5A7 subcutaneous administration every 3 days, the percentage of platelets negative for surface GPIbα expression was monitored by FACS.
Discussion
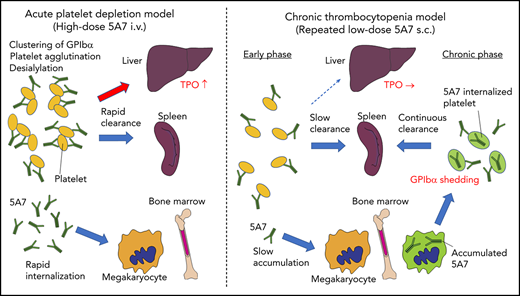
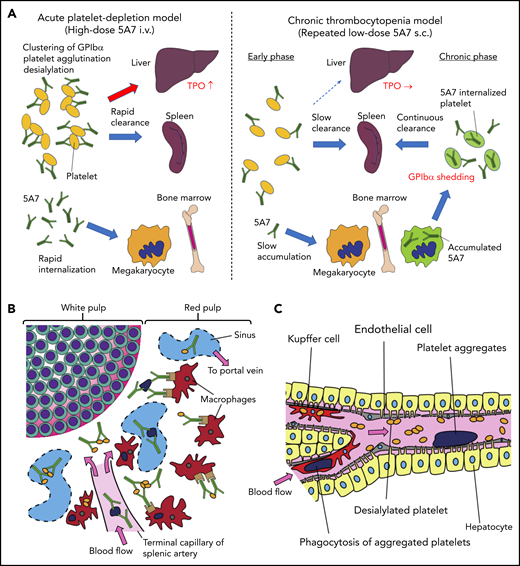
We have studied 2 mouse models of antibody-mediated thrombocytopenia using rat anti-mouse GPIbα mAb. Single IV administration of high-dose 5A7 caused rapid platelet depletion accompanied by TPO mRNA upregulation in the liver. Histological analysis showed sequestration of aggregated platelets in the liver and spleen at an early time point (30 minutes) after the antibody injection (Figure 6). Observation of liver sections at higher magnification shows the localization of a portion of opsonized platelets adjacent to hepatocytes as stained with anti-ASGPR1 (Figure 1D; supplemental Figure 3A-B). These results indicate that in this acute platelet-depletion model induced by 5A7, a part of opsonized platelets is cleared in the liver and signaling through AMR leads to TPO mRNA upregulation. It has been reported that an anti-GPIbα antibody blocked platelet-mediated hepatic TPO mRNA generation using a hepatic cell line.8 This contrasting result may be explained by the difference in the timing of TPO mRNA measurement, since the researchers incubated the hepatocyte cell line with mouse platelets for 24 hours before mRNA analysis. Alternatively, epitope specificities and agonistic activities of the antibodies, as well as cytokines secreted from activated platelets, may have modulatory effects on hepatic TPO generation. In our study, upregulation of TPO at early time points after IV antibody injection could be observed, not only with 5A7, but also with R300, a commercially obtained antibody. The platelet recovery was faster with R300 compared with 5A7, which may be related to the difference in the epitopes of these antibodies. The antibody 5A7 inhibits platelet binding to VWF A1 domain, and therefore, its epitope is thought to be at or close to the VWF binding region of GPIbα. The epitope of R300 is not known. An alternative explanation is that 5A7 is a rat mAb, whereas R300 is a mixture of multiple mAbs that may include multiple IgG subtypes. This difference may influence the half-life of the antibodies when administered in vivo.
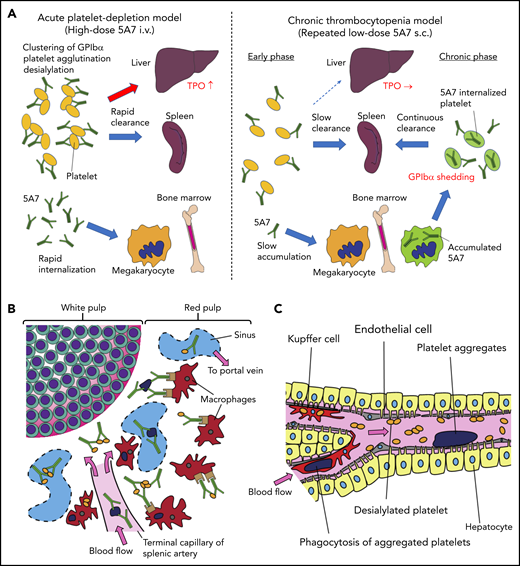
Schematic presentation showing different mechanisms of platelet clearance in 2 mouse models of antibody-induced thrombocytopenia. (A) In the acute platelet-depletion model (left), high-dose antibody causes platelet activation, aggregation, and desialylation. Aggregates and desialylated platelets are stuck in the microvasculature of the liver by binding to AMR. Opsonized platelets are also cleared by splenic macrophages via Fc receptor. Excess antibody is accumulated in MK in BM, which induces internalization of GPIbα. In the chronic thrombocytopenia model (right), 5A7 accumulates in MK in the BM and surface expression of GPIbα of newly produced platelets is decreased by shedding and internalization. Antibody concentration in plasma is not high enough to induce platelet aggregation, and thus, opsonized platelets are primarily cleared by splenic macrophages. (B) In the spleen, the antibody-bound platelets or platelet aggregates are cleared by macrophages via Fc receptor in both models. (C) In the acute platelet-depletion model, platelets are desialylated and aggregated. These platelets interact with hepatocytes through fenestrations in liver sinusoidal endothelial cells, transduce signal through AMR to upregulate TPO mRNA expression. Trapped desialylated platelets and aggregates are cooperatively cleared via hepatocyte capture and Kupffer cell phagocytosis. C-type lectins expressed on Kupffer cells, such as CLEC4F and macrophage galactose-type lectin,23,24 which have high affinity for desialylated glycoproteins, may contribute to this process.
Schematic presentation showing different mechanisms of platelet clearance in 2 mouse models of antibody-induced thrombocytopenia. (A) In the acute platelet-depletion model (left), high-dose antibody causes platelet activation, aggregation, and desialylation. Aggregates and desialylated platelets are stuck in the microvasculature of the liver by binding to AMR. Opsonized platelets are also cleared by splenic macrophages via Fc receptor. Excess antibody is accumulated in MK in BM, which induces internalization of GPIbα. In the chronic thrombocytopenia model (right), 5A7 accumulates in MK in the BM and surface expression of GPIbα of newly produced platelets is decreased by shedding and internalization. Antibody concentration in plasma is not high enough to induce platelet aggregation, and thus, opsonized platelets are primarily cleared by splenic macrophages. (B) In the spleen, the antibody-bound platelets or platelet aggregates are cleared by macrophages via Fc receptor in both models. (C) In the acute platelet-depletion model, platelets are desialylated and aggregated. These platelets interact with hepatocytes through fenestrations in liver sinusoidal endothelial cells, transduce signal through AMR to upregulate TPO mRNA expression. Trapped desialylated platelets and aggregates are cooperatively cleared via hepatocyte capture and Kupffer cell phagocytosis. C-type lectins expressed on Kupffer cells, such as CLEC4F and macrophage galactose-type lectin,23,24 which have high affinity for desialylated glycoproteins, may contribute to this process.
Interestingly, hepatic sequestration of opsonized platelets and TPO upregulation were not observed with our low-dose 5A7 subcutaneous injection model at any time points tested, and opsonized platelets were cleared by splenic macrophages, suggesting a difference in the mechanism of platelet clearance between these 2 models, possibly because of differences in the plasma 5A7 concentrations achieved (supplemental Figure 6). In patients with ITP, TPO upregulation is not generally observed, and it is often difficult to detect the autoantibody in plasma. Thus, despite the frequent use of high-dose antiplatelet IV injection for the studies of ITP, our study indicates that a low-dose subcutaneous injection model may better represent the status of patients with ITP. High doses of plasma antibody may be triggered by infection, altered immune homeostasis, or in specific situations such as posttransfusion purpura.21 In the acute platelet-depletion model, opsonized platelets observed in the liver mostly formed large aggregates (Figure 1D), indicating that high-dose 5A7 induces aggregation rather than GPIbα shedding, and the aggregated platelets are mainly trapped by the liver microvasculature. It is speculated that these trapped aggregates, as well as desialylated platelets, interact with hepatocytes through fenestrations in liver sinusoidal endothelial cells, transduce signal through AMR, and are cooperatively cleared via hepatocyte capture and Kupffer cell phagocytosis (Figure 6C).22,23 Several lectin receptors have been reported in the binding of hyposialylated platelets and as having a possible role in platelet clearance. CLEC4F, a member of C-type lectin expressed on Kupffer cells, has been demonstrated for binding to O-glycan–deficient platelets and their clearance in cooperation with hepatic AMR.23 More recently, macrophage galactose-type lectin was reported as a novel lectin receptor for hyposialylated VWF in addition to AMR on hepatycytes.24 Similarly, lectin receptors expressed on Kupffer cells may recognize desialylation of GPIbα O-glycans and contribute to cooperative clearance of opsonized platelets by hepatocyte and Kupffer cells. Further studies are needed to clarify this point. Subcutaneous administration of low-dose 5A7, on the other hand, did not achieve plasma 5A7 concentrations that were high enough to form large platelet aggregates or induce desialylation. In addition, platelets subsequently generated from 5A7-targeted MKs reduced GPIbα on their surface, by shedding and internalization. Therefore, newly produced platelets were primarily cleared in the spleen by F4/80+ cells, and hepatic TPO upregulation was not observed (Figure 6A). This is in agreement with what was previously reported in GPIbα-knockout mice.8
In patients with ITP, resistance to IVIG treatment has been reported to be more frequent when ITP is caused by anti-GPIb/IX than anti-GPIIb-IIIa.4,5 A signaling mechanism of GPIb-IX activation induced specifically by antibodies against the ligand-binding domain of GPIbα has also been reported.16 Consistent with these findings, mouse studies have identified anti-platelet antibody specificities as the determinant of FcR-dependent or FcR-independent clearance,7 thereby influencing the efficacy of IVIG treatment. However, a clinical study showed that antibody specificity was not the only determinant of the location of platelet clearance, either by the spleen or liver, or outcomes after splenectomy.25 Taken together, our results indicate that it is not only the antibody specificity but also plasma antibody dose that influence the locus of platelet clearance, TPO regulation, and responsiveness to IVIG.
Another key observation was that injected 5A7 not only bound to platelet GPIbα but also targeted GPIbα in BM MKs and was internalized. Our repeated subcutaneous injection model enabled us to analyze how 5A7 targeting MKs influences newly generated platelets. In this model, 5A7 is gradually accumulated in MKs in BM. Cleavage and internalization of 5A7-targeted GPIbα initiates in MKs, and therefore, newly generated platelets present with reduced GPIbα on their surface. This effect may explain, at least in part, the low sensitivity of diagnostic assays aimed at detecting autoantibodies bound to platelet surface glycoproteins in patients with suspected ITP.26 Previous efforts have been made to increase the sensitivity of the assays by detecting antibody-secreting B cells or internalized target proteins in platelets.27,28 However, so far, there is not a clear biomarker for ITP, and diagnosis is made by going through various analyses to exclude other causes of thrombocytopenia. Therefore, patients with other causes of thrombocytopenia are often misdiagnosed as ITP (or vice versa), resulting in unnecessary or improper treatment.29 Thus, generation of an assay that supports diagnosis of ITP will help reduce the risk of misdiagnosis. Of note, platelet GPIbα is more susceptible to cleavage in mice than in humans.17 Therefore, careful analysis of clinical samples is needed to confirm that measuring GPIbα expression of circulating platelets, corrected for size, would support the diagnosis of patients with ITP caused by anti-GPIbα autoantibodies.
For original data, e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Alessandro Zarpellon (The Scripps Research Institute, MERU-VasImmune Inc) for preparation of the 5A7 antibody and Jennifer Orje and Roberto Aiolfi (The Scripps Research Institute) for characterizing 5A7 by testing platelet adhesion under flow.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL-135290 (Z.M.R.) and HL-129011 (T.K.).
Authorship
Contributions: Y.M., E.W., and S.K. performed the experiments, interpreted the data, and helped in manuscript preparation; Z.M.R. provided vital reagents and reviewed and revised the manuscript; and T.K. designed the experiments, directed the study, and wrote the manuscript.
Conflict-of-interest disclosure: Z.M.R. is founder, president, and CEO of MERU-VasImmune Inc, which may develop commercial products based on methodologies presented in this article. S.K. and T.K. have equity interest in MERU-VasImmune Inc. The remaining authors declare no competing financial interests.
Correspondence: Taisuke Kanaji, IMM-210A, The Scripps Research Institute, 10550 North Torrey Pines Rd, La Jolla, CA 92037; e-mail: tkana@scripps.edu.