Key Points
Aberrant expression of CAR19+ in B-cell acute lymphoblastic leukemia blasts can be used as the target for αCAR19 T cells.
αCAR19 T cells also recognize CAR19+ T cells, therefore representing a potential strategy to deplete CART19 cells at long term.

Abstract
Unintentional transduction of B-cell acute lymphoblastic leukemia blasts during CART19 manufacturing can lead to CAR19+ leukemic cells (CARB19) that are resistant to CART19 killing. We developed an anti-CAR19 idiotype chimeric antigen receptor (αCAR19) to specifically recognize CAR19+ cells. αCAR19 CAR T cells efficiently lysed CARB19 cells in vitro and in a primary leukemia-derived xenograft model. We further showed that αCAR19-CART cells could be used as an “antidote” to deplete CART19 cells to reduce long-term side effects, such as B-cell aplasia.
Introduction
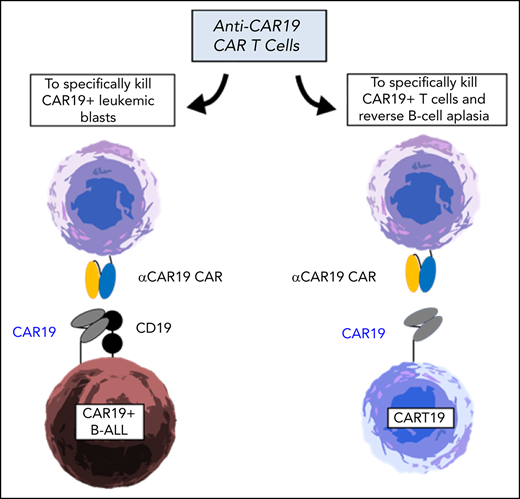
Anti-CD19 chimeric antigen receptor T cells (CART19) are now a standard therapeutic strategy for relapsed or refractory B-cell acute lymphoblastic leukemia (B-ALL).1 However, the high rate of complete responses is offset by a significant number of relapses, often with undetectable CD19 on the leukemic cells through several different mechanisms.2-5 Our group recently reported the extraordinary case of a pediatric patient with B-ALL treated with CART19 (CTL019) whose relapse blasts aberrantly expressed CAR19, the transgene intended for the T cells, due to unintentional transduction of a single leukemic cell with the CAR19 lentivirus during CTL019 manufacturing.6 This unique circumstance led to in cis binding between the CAR19 and CD19 on the cell surface, resulting in masking of the CD19 epitope and hence inability to be recognized by CART19 cells. Intriguingly, the aberrant integration of the CAR sequence into leukemic cells generates a specific tumor marker, CAR19, highly and homogeneously expressed on leukemic cells and absent from healthy tissues. We therefore hypothesized that targeting CAR19 on leukemic cells with a CAR will be an effective and safe strategy to treat CAR19+ leukemias. We developed a cellular product able to specifically target CAR19+ cells in the form of anti-CAR19 CAR T cells (αCAR19-CART) (Figure 1A). Moreover, we showed that this product could also serve as a depletion strategy for CAR19 T cells. CART19 depletion could thus be used in patients who were apparently cured of their underlying malignancy with CART19 and have prolonged B-cell aplasia and hypogammaglobulinemia due to the long-term persistence of CART19 cells.
![Anti-CAR19 CART cells to deplete CAR19+ leukemic cells. (A) Model: anti-CAR19 CART (αCAR19-CART) to recognize CAR19-expressing leukemic cells and trigger cytotoxicity. (B) Two novel CAR constructs were designed by using an anti-CAR19 scFv (clone 136.20.1) (two light chain orientations, H-L and L-H), a CD8 hinge, the 4-1BB costimulatory domain, CD3 zeta-signaling domain, and the pTRPE vector backbone. (C) The H-L and L-H αCAR19 CAR are efficiently expressed on T cells as detected by flow cytometry using a goat anti-human Ig Fab. (D) In vitro αCAR19-CART (L-H and H-L) efficiently kill CAR19+ B-ALL (NALM6) but not wild-type (WT) NALM6. The L-H construct exhibits a higher antileukemia effect and was therefore also tested in vivo. (E) NOD/SCID γ chain–deficient (NSG) mice were engrafted with luciferase-positive relapsed patient #107 leukemia blasts; then, at day 14, mice were randomized to receive no treatment, control T cells (untransduced [UTD]), CART19, CART22, or αCAR19-CART (1 × 106 cells/mouse). (F) CART19 and UTD cells are not able to control disease progression, but CART22 and αCAR19-CART L-H cells exhibit significant leukemia control. All experiments were repeated twice. (G) In an additional experiment, NSG mice were engrafted with luciferase-positive CAR19+ leukemia blasts obtained at relapse from patient #107; at day 14, mice were randomized to receive no treatment, UTD, αCAR19-CART H-L, or αCAR19-CART L-H. UTD cells are not able to control disease progression, but both αCAR19-CARTs, in particular L-H, show an advantage in overall survival. Survival curves were compared by using the log-rank test. Asterisks are used in each figure to represent P values (*P < .05, **P < .01, ****P < .0001).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/7/10.1182_blood.2019001859/5/m_bloodbld2019001859f1.png?Expires=1766130189&Signature=J2leNtwCVxZU8CZqv8rUCfGbdMp4s7Tm7SCULfsfQWw4EXnAzGQiR7f~L02e0cLnO1FWTF4H8ewn7P7M2Qx4MtI-tZWaMhUts6UHfuXWflh9EWM1tBVNKEg9airTdeQeQh9tMPXnOv~9mTF1y4qFflsvLJWKNxLvi9H6tGDiUJwAkHToRXSOlnjNctsOD3u56t4qAMZo0e36fNPks-u8B8xInjv4yLvgt2EEpq6LPSlD4WCpFqWyxrpeiL8X0Wuidfgc3TKOste6urSpLZZEczG6wBc3MKEisrC1iIVjZK1B0uM4t6G6hdDLQ1QYauzHBOMdSUEpzmnwzVBogwWuHg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Anti-CAR19 CART cells to deplete CAR19+ leukemic cells. (A) Model: anti-CAR19 CART (αCAR19-CART) to recognize CAR19-expressing leukemic cells and trigger cytotoxicity. (B) Two novel CAR constructs were designed by using an anti-CAR19 scFv (clone 136.20.1) (two light chain orientations, H-L and L-H), a CD8 hinge, the 4-1BB costimulatory domain, CD3 zeta-signaling domain, and the pTRPE vector backbone. (C) The H-L and L-H αCAR19 CAR are efficiently expressed on T cells as detected by flow cytometry using a goat anti-human Ig Fab. (D) In vitro αCAR19-CART (L-H and H-L) efficiently kill CAR19+ B-ALL (NALM6) but not wild-type (WT) NALM6. The L-H construct exhibits a higher antileukemia effect and was therefore also tested in vivo. (E) NOD/SCID γ chain–deficient (NSG) mice were engrafted with luciferase-positive relapsed patient #107 leukemia blasts; then, at day 14, mice were randomized to receive no treatment, control T cells (untransduced [UTD]), CART19, CART22, or αCAR19-CART (1 × 106 cells/mouse). (F) CART19 and UTD cells are not able to control disease progression, but CART22 and αCAR19-CART L-H cells exhibit significant leukemia control. All experiments were repeated twice. (G) In an additional experiment, NSG mice were engrafted with luciferase-positive CAR19+ leukemia blasts obtained at relapse from patient #107; at day 14, mice were randomized to receive no treatment, UTD, αCAR19-CART H-L, or αCAR19-CART L-H. UTD cells are not able to control disease progression, but both αCAR19-CARTs, in particular L-H, show an advantage in overall survival. Survival curves were compared by using the log-rank test. Asterisks are used in each figure to represent P values (*P < .05, **P < .01, ****P < .0001).
Anti-CAR19 CART cells to deplete CAR19+ leukemic cells. (A) Model: anti-CAR19 CART (αCAR19-CART) to recognize CAR19-expressing leukemic cells and trigger cytotoxicity. (B) Two novel CAR constructs were designed by using an anti-CAR19 scFv (clone 136.20.1) (two light chain orientations, H-L and L-H), a CD8 hinge, the 4-1BB costimulatory domain, CD3 zeta-signaling domain, and the pTRPE vector backbone. (C) The H-L and L-H αCAR19 CAR are efficiently expressed on T cells as detected by flow cytometry using a goat anti-human Ig Fab. (D) In vitro αCAR19-CART (L-H and H-L) efficiently kill CAR19+ B-ALL (NALM6) but not wild-type (WT) NALM6. The L-H construct exhibits a higher antileukemia effect and was therefore also tested in vivo. (E) NOD/SCID γ chain–deficient (NSG) mice were engrafted with luciferase-positive relapsed patient #107 leukemia blasts; then, at day 14, mice were randomized to receive no treatment, control T cells (untransduced [UTD]), CART19, CART22, or αCAR19-CART (1 × 106 cells/mouse). (F) CART19 and UTD cells are not able to control disease progression, but CART22 and αCAR19-CART L-H cells exhibit significant leukemia control. All experiments were repeated twice. (G) In an additional experiment, NSG mice were engrafted with luciferase-positive CAR19+ leukemia blasts obtained at relapse from patient #107; at day 14, mice were randomized to receive no treatment, UTD, αCAR19-CART H-L, or αCAR19-CART L-H. UTD cells are not able to control disease progression, but both αCAR19-CARTs, in particular L-H, show an advantage in overall survival. Survival curves were compared by using the log-rank test. Asterisks are used in each figure to represent P values (*P < .05, **P < .01, ****P < .0001).
Study design
Lentiviral constructs
The murine anti-CD19 CAR was generated as previously described.7,8 We designed 2 single-chain variable fragments (scFvs) able to specifically recognize CAR19 using the heavy and light chains (either H-L or L-H) from an anti-idiotype monoclonal antibody (clone 136.20.1).9 These scFvs were then cloned into the CTL019 CAR backbone10 (Figure 1B). The control anti-CD22 scFv was generated by using the m971 antibody11 and cloned into the CTL019 CAR backbone.12 The CAR33 and CAR123 constructs were previously published.13,14 The constructs were packaged into lentiviral vectors (pTRPE, a University of Pennsylvania proprietary lentiviral backbone) by using 293T cells.
Patient and healthy donor samples
Bone marrow cells from patient #107 at relapse were obtained at the clinical practices of the University of Pennsylvania/The Children’s Hospital of Philadelphia under an Institutional Review Board protocol and were used to generate the primary ALL xenograft model. The trial in which patient #107 participated was conducted at The Children’s Hospital of Philadelphia and has been previously reported (NCT01626495).15,16 The primary leukemia cells of patient #107 from relapse after CTL019 were expanded in nonobese diabetic/severe combined immunodeficiency (NOD/SCID) γ chain–deficient (NSG) mice.6 De-identified normal donor peripheral blood specimens or CD4+ and CD8+ peripheral T cells were obtained from the Human Immunology Core of the University of Pennsylvania. All subjects provided written informed consent according to the Declaration of Helsinki and the International Conference on Harmonisation Guidelines for Good Clinical Practice. All ethical regulations were followed. Production of CAR-expressing T cells for the research experiments was performed as previously described.17
Additional details are provided in the supplemental Methods (available on the Blood Web site).
Results and discussion
We designed a second-generation CAR using the 4-1BB and CD3z backbone and the heavy and light chain sequences of an anti-idiotype monoclonal antibody that is used to detect CAR19 (FMC63) with flow cytometry (clone 136.20.1, patent WO 2014/190273 Al)9 (Figure 1B). This antibody was previously shown to specifically recognize CAR19+ cells and not stain peripheral blood mononuclear cells and several cancer cell lines.9 We confirmed its specificity for CAR19 (FMC63) by testing it against an array of different CAR constructs, and only CAR19+ T cells were stained (supplemental Figure 1A). Both the L-H and H-L orientation of the anti-CAR19 CAR were efficiently expressed on T cells as detected by flow cytometry (Figure 1C; supplemental Figure 1B).
We then sought to test the ability of the αCAR19-CART to kill CAR19+ leukemic cells. To this end, we used a luciferase-positive B-ALL cell line (NALM6) engineered to express the same CAR19 construct used in clinical trials as target cells.6 As previously shown, expression of CAR19+ on NALM6 cells abrogated CD19 detection by flow cytometry and led to resistance to CART19 killing6 (Figure 1D). In luciferase-based killing assays, αCAR19-CART (L-H orientation, P < .0001; H-L orientation, P = .0063), but not CART19, efficiently killed CAR19-expressing B-ALL (NALM6) but not wild-type NALM6 (P = not significant). The L-H–oriented construct was superior to the H-L–oriented construct (P = .0073). We therefore selected the L-H αCAR19-CART for further validation.
NOD-SCID γ chain–deficient mice were engrafted with primary CAR19+ leukemic blasts derived from patient #1076 that had been previously passaged in mice and transduced with luciferase for detection (Figure 1E). At day 14, mice were randomized to a group based on tumor burden (luminescence) to receive no treatment, control T cells (UTD), CART19, CART22, or αCAR19-CART L-H at an equal number of CAR-expressing cells (1 × 106 cells/mouse). For these experiments, a suboptimal (not curative) dose of T cells was used to stress the differences between the different treatments. As expected, CART19 and UTD failed to control disease progression, whereas αCAR19-CART L-H exhibited significant leukemia control (P < .0001) (Figure 1F), and this control was even better than that of CART22. In a repeated experiment including also the H-L orientation of the αCAR19-CART, we injected 3 × 106 CAR+ T cells per mouse and obtained significant improvement in overall survival with both L-H and H-L, with 100% of mice treated with αCAR19-CART L-H alive at long term (Figure 1G). This anti-leukemia effect was associated with significant αCAR19-CART expansion in the peripheral blood (supplemental Figure 2A-B).
Having shown that αCAR19-CART can efficiently target CAR19+ leukemia in vitro and in vivo, we sought to validate another possible use for this product: a strategy to deplete CART19 cells in patients who have achieved complete remission after CART19 treatment (Figure 2A) and have continuous B-cell aplasia due to long-term persistence of CART19. To this end, we performed killing assays coculturing CART19 and αCAR19-CART (Figure 2B). CART22 was used as a control. At 48 hours, both H-L and L-H αCAR19-CART cells were able to kill CART19 but not CART22 or control T cells. We speculated that as αCAR19-CART can kill CART19, CART19 could theoretically also cause αCAR19-CART death as a form of bidirectional killing. To model this hypothesis, we cocultured αCAR19-CART with CART19 cells at different effector-to-target ratios and evaluated tumor killing at 24 and 48 hours. Interestingly, although CART19 were killed by αCAR19-CART (H-L or H-L), there was no evidence of a reverse killing of αCAR19-CART cells by CART19 cells (Figure 2C).
![Anti-CAR19 CART cells deplete CAR19+ T cells. (A) Model: anti-CAR19 CART (αCAR19-CART) can recognize CAR19-expressing T cells and trigger cytotoxicity. (B) αCAR19-CART H-L (left) or L-H (right) (effectors) were cocultured with control T cells, CART19, or CART22 cells (targets, CellTracker Orange [CTV-positive]) and the percentage of surviving target cells was measured at 48 hours using flow cytometry. CART19 cells were specifically killed by αCAR19-CART. (C) αCAR19-CART H-L (left) or L-H (right) (effectors, carboxyfluorescein succinimidyl ester–positive) were cocultured with control T cells, CART19, or CART22 cells (targets, CTV-positive) at different effector-to-target (E:T) ratios, and changes on the E:T ratios were recorded at 48 hours. For E:T ratios >2, αCAR19-CART killing prevails over CART19. A total of 100 000 events were acquired for each condition. All experiments were repeated twice. Asterisks are used in each figure to represent P values (*P < .05, **P < .01).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/7/10.1182_blood.2019001859/5/m_bloodbld2019001859f2.png?Expires=1766130189&Signature=Qkjpw~3cYxYUmqcfQkCrGHj4j9AnIS8wxVv2z4C7h70oTudRk~ZV9-Fukyhi7gwClfG4P1PuRofuYfIMHu0f~RcYpuprcKkXn~89NQuOsraIsdhcWs3RtNI~AczlygNqWuFBhaD1D7F~1NKzYFoDwBKhLX7N5LV38FF-v3zfqoYR-iXarT7GvtRycsReCJvSKJVekcs70TemGHplNQjMzvuJ3Ovrl8uGfQCKouZ6EQAGHJkIFEDRguQ3gkEjrNPHpH2vUWQ5DBYmv49Y5N-62rHLWFzg7fBoaE-QwfwJCctvOmQdORAUT2QXTlMWrsHjLLULPoWxIMRxPXDZwEm3JA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Anti-CAR19 CART cells deplete CAR19+ T cells. (A) Model: anti-CAR19 CART (αCAR19-CART) can recognize CAR19-expressing T cells and trigger cytotoxicity. (B) αCAR19-CART H-L (left) or L-H (right) (effectors) were cocultured with control T cells, CART19, or CART22 cells (targets, CellTracker Orange [CTV-positive]) and the percentage of surviving target cells was measured at 48 hours using flow cytometry. CART19 cells were specifically killed by αCAR19-CART. (C) αCAR19-CART H-L (left) or L-H (right) (effectors, carboxyfluorescein succinimidyl ester–positive) were cocultured with control T cells, CART19, or CART22 cells (targets, CTV-positive) at different effector-to-target (E:T) ratios, and changes on the E:T ratios were recorded at 48 hours. For E:T ratios >2, αCAR19-CART killing prevails over CART19. A total of 100 000 events were acquired for each condition. All experiments were repeated twice. Asterisks are used in each figure to represent P values (*P < .05, **P < .01).
Anti-CAR19 CART cells deplete CAR19+ T cells. (A) Model: anti-CAR19 CART (αCAR19-CART) can recognize CAR19-expressing T cells and trigger cytotoxicity. (B) αCAR19-CART H-L (left) or L-H (right) (effectors) were cocultured with control T cells, CART19, or CART22 cells (targets, CellTracker Orange [CTV-positive]) and the percentage of surviving target cells was measured at 48 hours using flow cytometry. CART19 cells were specifically killed by αCAR19-CART. (C) αCAR19-CART H-L (left) or L-H (right) (effectors, carboxyfluorescein succinimidyl ester–positive) were cocultured with control T cells, CART19, or CART22 cells (targets, CTV-positive) at different effector-to-target (E:T) ratios, and changes on the E:T ratios were recorded at 48 hours. For E:T ratios >2, αCAR19-CART killing prevails over CART19. A total of 100 000 events were acquired for each condition. All experiments were repeated twice. Asterisks are used in each figure to represent P values (*P < .05, **P < .01).
Thus, the acquisition of CAR expression by leukemic cells during CART manufacturing is an extremely rare event that can cause resistance to CAR T cells targeting CD19 and CD22.6 This event generates a tumor-specific antigen, thereby presenting an opportunity for specific targeting without off-target toxicity. In this context, we showed that an αCAR19-CART product efficiently targets CAR19+ B-ALL in vitro and in vivo.
The first pediatric patient treated with the CART19 product that is now known as tisagenlecleucel is currently in her seventh year of remission; she continues to have B-cell aplasia and requires frequent immunoglobulin infusions to prevent infections. Several other pediatric and adult patients are in a similar situation today and more will be in the next few years thanks to the widespread use of CART19 in the routine clinical practice. We showed that αCAR19-CART can specifically deplete CAR19 T cells, potentially eliminating the common side effect of prolonged B-cell aplasia or possibly other as-yet unknown complications associated with long-term CAR T-cell engraftment.18 Importantly, B-cell aplasia and hypogammaglobulinemia also occur in adult patients with lymphoma treated with anti-CD19 CAR T cells and, therefore, this product may also be useful in such patients. The inclusion of suicide genes in future CART products could be an effective strategy to deplete CAR19+ B-ALL blasts and also T cells. However, the 2 most advanced CART19 products (tisagenlecleucel19 and axicabtagene ciloleucel20 ) do not include safety switches such as iCaspase921 or endothelial growth factor receptor expression.22 Moreover, the effectiveness of safety switches in the context of CART therapy has yet to be proven in the clinic, and preclinical results show that an iCaspase9-resistant T-cell population can emerge.
In conclusion, with the exponential growth of the use of tisagenlecleucel in pediatric and young adult patients, a significant number of patients will be in long-term remission but at risk of infections and constant need of medical care. In this context, such an approach could address a future significant medical need.
All raw and analyzed data are promptly available upon request. Data access requests will be evaluated by the University of Pennsylvania Center for Innovation, who manages the intellectual property of this project.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The anti-CAR19 heavy and light chain sequence (clone 136.20.1) was originally obtained from patent WO2014190273A1 and subsequently modified in a CAR scFv. One of the CARs used in this study was obtained under a material transfer agreement from Dario Campana, Chihaya Imai, and St. Jude Children’s Research Hospital and was subsequently modified by cloning into a lentiviral vector and expressed with a eukaryotic promoter.23
This work was supported by grants from the University of Pennsylvania–Novartis Alliance (Principal Investigators [PIs]: C.H.J., S.I.G., and M.R.), the EMD Serono Cancer Immunotherapy Clinical Fellowship by the Society for Immunotherapy of Cancer (PI: M.R.), the Bristol-Myers Squibb Oncology Fellowship in Clinical Cancer Research by the American Association for Cancer Research (PI: M.R.), Gabrielle’s Angel Foundation (PIs: M.R. and D.M.B.), the SIES-AIL fellowship by the Italian Society of Experimental Hematology and the Italian Leukemia Association (PI: M.R.), the ASH Scholar Award (PI: M.R.), the National Institutes of Health, National Cancer Institute (1K99CA212302-01A1 and R00CA212302) (PI: M.R.), and the St. Baldrick’s Foundation Scholar Award (PI: D.M.B.).
Authorship
Contribution: M.R. formulated the initial idea and planned the experiments; M.R., D.M.B., and O.S. performed the experiments, analyzed the data, and contributed to the manuscript; M.K. and A.D.P. designed the plasmids; D.M.B., J.P., S.J.H., O.S., and M.R. performed the animal experiments; M.R. wrote the manuscript; S.I.G., J.J.M., S.F.L., and C.H.J. edited the manuscript; and all authors reviewed and accepted the contents of the article.
Conflict-of-interest disclosure: C.H.J., M.R., J.J.M., S.F.L., A.D.P., and S.I.G. are inventors of intellectual property in the field of CAR immunotherapy. D.M.B. discloses a consultant relationship with Eureka Therapeutics. M.R. discloses a consultant relationship with NanoString Inc. and AbClon. The remaining authors declare no competing financial interests.
Correspondence: Marco Ruella, Center for Cellular Immunotherapies, Perelman School of Medicine at the University of Pennsylvania, 3400 Civic Center Blvd, Perelman Center for Advanced Medicine, South Pavillion Extension, 8-112, Philadelphia, PA 19104; e-mail: mruella@upenn.edu.