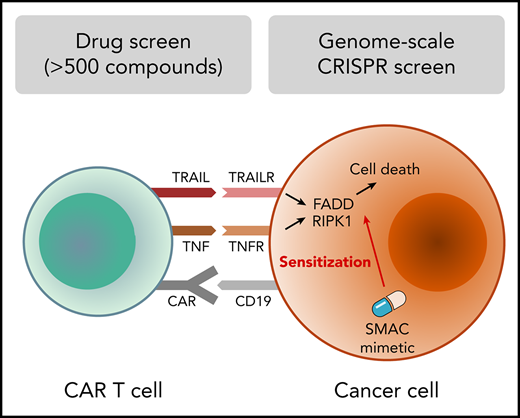
Key Points
Survey of immunomodulatory effects of >500 drugs identifies SMAC mimetics as sensitizers to CAR T-cell cytotoxicity.
Genome-scale CRISPR screen reveals essentiality of death receptor signaling for CAR T-cell cytotoxicity.

Abstract
Chimeric antigen receptor (CAR) T-cell therapy has proven effective in relapsed and refractory B-cell malignancies, but resistance and relapses still occur. Better understanding of mechanisms influencing CAR T-cell cytotoxicity and the potential for modulation using small-molecule drugs could improve current immunotherapies. Here, we systematically investigated druggable mechanisms of CAR T-cell cytotoxicity using >500 small-molecule drugs and genome-scale CRISPR-Cas9 loss-of-function screens. We identified several tyrosine kinase inhibitors that inhibit CAR T-cell cytotoxicity by impairing T-cell signaling transcriptional activity. In contrast, the apoptotic modulator drugs SMAC mimetics sensitized B-cell acute lymphoblastic leukemia and diffuse large B-cell lymphoma cells to anti-CD19 CAR T cells. CRISPR screens identified death receptor signaling through FADD and TNFRSF10B (TRAIL-R2) as a key mediator of CAR T-cell cytotoxicity and elucidated the RIPK1-dependent mechanism of sensitization by SMAC mimetics. Death receptor expression varied across genetic subtypes of B-cell malignancies, suggesting a link between mechanisms of CAR T-cell cytotoxicity and cancer genetics. These results implicate death receptor signaling as an important mediator of cancer cell sensitivity to CAR T-cell cytotoxicity, with potential for pharmacological targeting to enhance cancer immunotherapy. The screening data provide a resource of immunomodulatory properties of cancer drugs and genetic mechanisms influencing CAR T-cell cytotoxicity.
Introduction
Chimeric antigen receptor (CAR) T-cell therapy has induced remarkable responses in relapsed and refractory B-cell malignancies.1-4 However, both primary and adaptive resistance to CAR T-cell therapy occur.5-9 Existing oncological and other drugs could be harnessed to improve CAR T-cell therapy and other immunotherapies.10 Targeted small-molecule inhibitors may have unexpected immunomodulatory properties as a result of inhibition of pathways regulating immune functions that may overlap with oncogenic processes.11 Systematic understanding of these immunomodulatory functions of drugs would facilitate prioritizing combination candidates for immunotherapies.
Stimulation of T cells through the T-cell receptor (TCR) or CAR result in the activation and/or nuclear translocation of several key transcription factors (TFs), including NFAT, AP-1, and NF-κB, which orchestrate multiple T-cell responses, including proliferation, cytokine production, and effector functions.12 The cytotoxic function of CD8+ T lymphocytes is essential for immunotherapies relying on T cells. Cytotoxicity can occur through the release of cytolytic granule contents, including granzymes and perforin, or alternatively by exposure of death receptor ligands, such as FASL, TRAIL, and TNF.13 Drugs and genetic alterations influencing these mechanisms of T-cell activation and cytotoxicity may affect the efficacy of T cell–based immunotherapies.
Large-scale drug sensitivity screens in cancer cells have comprehensively mapped the pharmacogenomic interactions in various cancer types14-16 and, more recently, the effects of small molecules on cell-cell contact interactions of leukocytes.17 However, no efforts to systematically map combination therapy candidates for CAR T-cell therapy have been made at scale. The cancer cell–intrinsic molecular mechanisms underlying sensitivity or resistance to TCR-dependent T-cell killing have been investigated on a genomic scale using CRISPR-Cas9 loss-of-function screens,18-21 but similar understanding is lacking for CAR T cells.
Here, we approached these questions by integrating pharmacological profiling and genome-scale CRISPR-mediated genetic perturbation screens in coculture models using anti-CD19 CAR T cells and cancer cells as well as TCR signaling TF reporter assays. Our study provides a resource for immunomodulatory properties of cancer drugs influencing cytotoxic T-cell interactions and T-cell activation.
Methods
More detailed descriptions of the experimental methods and analyses are available in supplemental Methods.
Cell lines and CAR T cells
The NALM6 cell line was obtained from Olli Lohi (University of Tampere, Tampere, Finland); 697, KASUMI2, RCHACV, and TOM1 lines were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH; and DB, RIVA, and SUDHL4 lines were kindly provided by Sirpa Leppä (University of Helsinki, Helsinki, Finland). The Jurkat triple parameter reporter cell line containing NFAT-EGFP, NF-κB-ECFP, and AP-1-mCherry reporters,22 the Jurkat-AP-1-EGFP reporter cell line,23 and the T-cell stimulator (TCS) cells and TCS-CD80 cells engineered to constitutively express CD8024 were obtained from Peter Steinberger. CAR T cells were manufactured as previously described.25 The cell lines were transduced to express luciferase or Cas9 as described in the supplemental Methods.
Patients
For primary patient cell cytotoxicity assays, we used viably frozen diagnostic-phase bone marrow mononuclear cells from adult precursor B-cell acute lymphoblastic leukemia (B-ALL) patients (n = 18). The samples were obtained from the Finnish Hematology Registry and Clinical Biobank with appropriate ethics approval (Dnro 202/06.01.00/2013, Helsinki University Hospital Ethics Committee) in accordance with the Declaration of Helsinki.
CAR T-cell coculture drug sensitivity screens
The oncology compound collection included 165 US Food and Drug Administration/European Medicines Agency–approved anticancer and other drugs and 361 investigational and preclinical compounds (supplemental Table 1). The coculture screens were performed using an adaptation of the drug sensitivity and resistance testing platform described previously.15 Briefly, target cells were incubated either alone or with CAR T cells in 384-well plates containing the compounds in 5 increasing concentrations covering a 10 000-fold concentration range for 24 hours, after which the target cell viability was measured using luciferase assay. Drug responses were quantified using the drug sensitivity score (DSS).26 The effect of drugs on CAR T-cell cytotoxicity was measured as differential DSS between target cells treated with CAR T cells and target cells only.
TCR signaling TF reporter drug sensitivity screens
Jurkat triple parameter reporter cells were used for NFAT and NF-κB screens and Jurkat-AP-1-EGFP cells for AP-1 screens. Jurkat reporter cells cocultured with TCS or TCS-CD80 cells were exposed to the drug library for 24 hours, after which reporter activity was measured using an iQue Screener Plus flow cytometer (Intellicyt).
Flow cytometry–based primary B-ALL cytotoxicity assay
Frozen bone marrow mononuclear cells of B-ALL patients were thawed and cocultured on a 96-well plate with or without CAR T cells in the presence of birinapant or dimethyl sulfoxide (DMSO). After 24 hours, cells were stained with antibodies for CD8, CD4, CD3, PD-1, CD19, and CD69 and the viability markers annexin V and 7-AAD. Data were acquired using an iQue Screener Plus flow cytometer and analyzed using ForeCyt software (edition 6.2; Intellicyt) to obtain counts of viable cells in the CD19+ population.
Genome-wide CRISPR-Cas9 screens
NALM6-Cas9 cells were transduced with the GeCKO v2 library27,28 and selected with puromycin for 6 days starting 48 hours posttransduction. On day 8 posttransduction, cells were divided into the following conditions: NALM6 only, CAR T cells plus NALM6 at a 1:35 effector/target ratio, and CAR T cells plus 10 nM of birinapant plus NALM6 at a 1:70 effector/target ratio. Cells were pelleted after 9 to 12 days from the start of the screen, frozen at −70°C, and later thawed for genomic DNA extraction using the Blood Maxi Kit (Qiagen). Amplicons containing single-guide RNA (sgRNA) sequences were amplified with a 2-step PCR protocol (supplemental Table 8) and sequenced using a HiSeq 2000 (Illumina). Data were analyzed using MAGeCK29 with default parameters.
CRISPR-Cas9 screen hit validation
Individual sgRNAs targeting genome-wide screen hit genes or control sgRNAs in the lentiCRISPRv2 plasmid (a gift from Feng Zhang; Addgene #52961) were lentivirally introduced into luciferase-expressing NALM6, RCHACV, and SUDHL4 cells. Editing efficiency was assessed using deep sequencing and CRISPResso230 or Sanger sequencing and TIDE31 (supplemental Tables 9-10) and flow cytometry. Target cells harboring each sgRNA were cultured alone or with CAR T cells or control T cells in indicated effector/target ratios, with 100 nM of birinapant or DMSO for 24 hours, after which target cell viability was measured using luciferase assay. Raw luminescence values were normalized to the average of technical replicates of DMSO-treated target cells carrying each sgRNA.
Differential gene expression analysis and correlations
Hypergeometric test followed by Bonferroni adjustment of P values was used to estimate statistical enrichment of gene expression in a cancer type within Hemap32 B-cell cancers. Two-tailed Wilcoxon rank sum test followed by Benjamini-Hochberg adjustment of P values to obtain false discovery rates (FDRs) was used to estimate differential gene expression in samples of a particular genetic subgroup of B-ALL compared with all the other samples. For genomic and clinical correlations with gene expression in diffuse large B-cell lymphoma (DLBCL), different feature types (gene expression, clinical, copy-number variation, mutations, and sample annotation) were correlated with death receptor gene expression using Spearman correlation followed by Benjamini-Hochberg adjustment of P values.
Statistical analysis
The statistical details of all experiments are reported in the text, figure legends, and figures, including statistical analyses performed, statistical significance, and sample counts. In boxplots, the horizontal line indicates the median, boxes indicate the interquartile range, and whiskers extend from the hinge to the smallest/largest value, at most 1.5 × interquartile range from the hinge.
Results
A coculture screen for drugs modulating interactions between CAR T cells and cancer cells
To identify small-molecule drugs influencing CAR T-cell cytotoxicity, we carried out a high-throughput drug screen using a coculture assay with CD19-directed CAR T cells harboring CD28 and CD3ζ signaling domains and CD19+ NALM6 B-ALL cells expressing luciferase (NALM6-luc cells; (Figure 1A; supplemental Figure 1). An increasing effector/target ratio of CD19 CAR T cells to NALM6-luc cells led to dose-dependent reduction of luminescence, whereas no change was observed with empty vector–transduced T cells, demonstrating that the 384-well format luciferase assay accurately monitors target cell viability without interfering signals from T cells (Figure 1B). In the drug screen, we used a library of 526 approved or investigational compounds spanning several functional classes, including conventional chemotherapy agents, kinase inhibitors, apoptotic modulators, and epigenetic and metabolic modifiers, as well as several nononcology drugs (Figure 1A; supplemental Table 1). We exposed NALM6-luc cells to the compounds at 5 different concentrations for 24 hours both alone and in the presence of CAR T cells and measured specific target cell viability using the luciferase assay (Figure 1A). To compare drug responses between CAR T cell–treated and control NALM6-luc cells, we calculated percent inhibition values at each dose based on luminescence readouts and summarized the overall responses using the differential DSS26 based on the area between the dose-response curves of the 2 conditions (individual drug response curves are shown in supplemental Table 1).
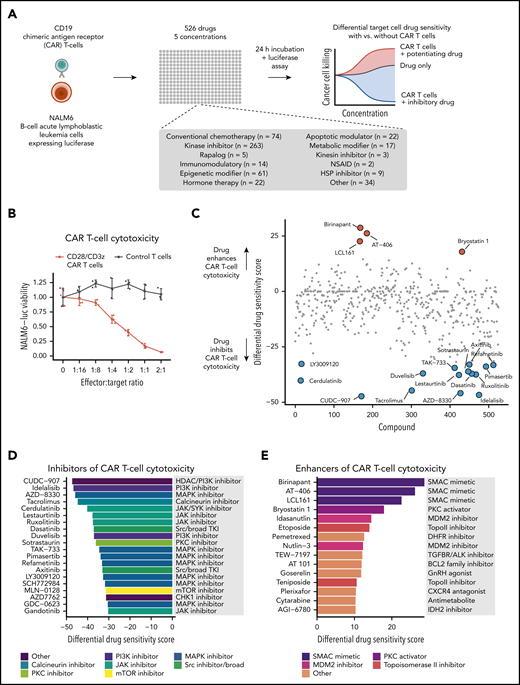
High-throughput drug screen to identify drugs modulating CAR T-cell cytotoxicity. (A) Schematic of the high-throughput coculture system drug sensitivity screen. (B) NALM6-luc cell viability with different effector/target ratios of CAR T cells or empty vector–transduced control T cells and NALM6-luc cells cocultured for 24 hours. (C) Overview of drug responses in CAR T-cell cytotoxicity screen. A positive differential DSS between CAR T cell–treated and control screens indicates that the compound enhances CAR T-cell cytotoxicity, whereas a negative score indicates inhibition. (D) Top 20 drugs most potently inhibiting CAR T-cell cytotoxicity ordered by the differential DSS. (E) Top 15 drugs most potently enhancing CAR T-cell cytotoxicity ordered by the differential DSS. NSAID, nonsteroidal anti-inflammatory drug.
High-throughput drug screen to identify drugs modulating CAR T-cell cytotoxicity. (A) Schematic of the high-throughput coculture system drug sensitivity screen. (B) NALM6-luc cell viability with different effector/target ratios of CAR T cells or empty vector–transduced control T cells and NALM6-luc cells cocultured for 24 hours. (C) Overview of drug responses in CAR T-cell cytotoxicity screen. A positive differential DSS between CAR T cell–treated and control screens indicates that the compound enhances CAR T-cell cytotoxicity, whereas a negative score indicates inhibition. (D) Top 20 drugs most potently inhibiting CAR T-cell cytotoxicity ordered by the differential DSS. (E) Top 15 drugs most potently enhancing CAR T-cell cytotoxicity ordered by the differential DSS. NSAID, nonsteroidal anti-inflammatory drug.
We identified several compounds that strongly inhibited CAR T-cell cytotoxicity (Figure 1C-D). The calcineurin inhibitor tacrolimus, an immunosuppressant used to prevent graft rejection, was among the top inhibitory compounds, confirming that the screening approach is able to recover biologically relevant hits. Other inhibitors of CAR T-cell cytotoxicity included tyrosine kinase inhibitors (TKIs) targeting the MAPK pathway (pimasertib and refametinib), JAK (ruxolitinib, lestaurtinib, and gandotinib), and PI3K (idelalisib and duvelisib), as well as broad-spectrum kinase inhibitors inhibiting SRC, among other targets (dasatinib and axitinib).
Strikingly, the 3 drugs that most potently enhanced CAR T-cell cytotoxicity (birinapant, AT-406, and LCL-161) all belong to the same drug class of SMAC mimetics or inhibitor of apoptotic protein (IAP) antagonists33 (Figure 1C,E). Other compounds that enhanced lysis of NALM6-luc cells by CAR T cells included the PKC activator bryostatin-1, MDM2 inhibitors (idasanutlin and nutlin-3), and topoisomerase 2 inhibitors (etoposide and teniposide).
MAPK pathway and SRC inhibitors impair CAR T-cell cytotoxicity through TCR signaling inhibition
Several mechanisms may mediate the inhibition of CAR T-cell cytotoxicity by small molecules, such as suppression of TCR signaling downstream of the CD28/CD3ζ CAR. To investigate the contribution of TCR signaling impairment to the inhibitory effects on CAR T-cell cytotoxicity, we screened the same drug library using Jurkat T-cell lines expressing fluorescent reporters for the TCR signaling TFs NFAT, NF-κB, and AP-122 with or without CD28-mediated costimulation (Figure 2A; supplemental Figure 2; supplemental Tables 2-3).
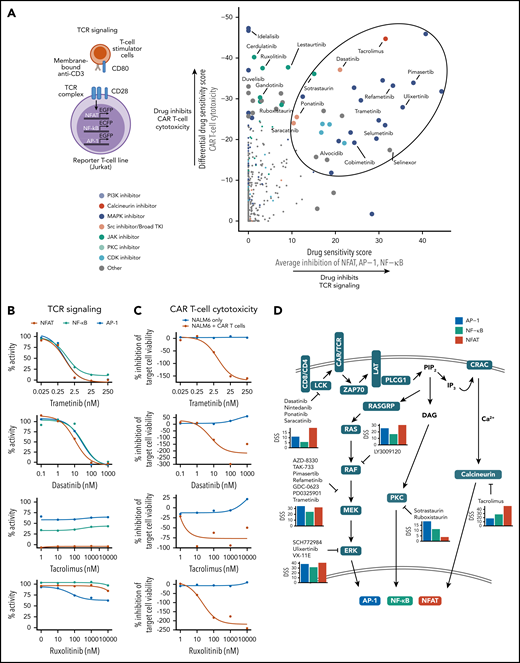
Suppression of CAR T-cell cytotoxicity through TCR signaling inhibition. (A) Schematic of the TCR signaling reporter system is shown on the left. Scatter plot comparing drug-mediated inhibition of CAR T-cell cytotoxicity and average inhibition of NFAT, AP-1, and NF-κB reporter activity quantified by the DSS is shown on the right. Data are from screens with CD28 costimulation. Enlarged points indicate drugs with T-cell reporter DSS >15 or CAR T-cell cytotoxicity DSS below −25, with selected drugs labeled, and drugs with high inhibition of both cytotoxicity and TCR signaling circled. (B) Dose-response curves of NFAT, NF-κB, and AP-1 reporter activity for selected drugs. Data are from screens with CD28 costimulation. (C) Dose-response curves of CAR T-cell cytotoxicity for selected drugs. (D) Schematic of proposed mechanisms of CAR T-cell cytotoxicity and TCR signaling inhibition by selected drugs of 50 most potent CAR T-cell cytotoxicity inhibitors. Average DSSs of AP-1 (blue), NF-kB (green), and NFAT (red) inhibition for the listed drugs in the CD28 costimulation screens are shown as bar plots.
Suppression of CAR T-cell cytotoxicity through TCR signaling inhibition. (A) Schematic of the TCR signaling reporter system is shown on the left. Scatter plot comparing drug-mediated inhibition of CAR T-cell cytotoxicity and average inhibition of NFAT, AP-1, and NF-κB reporter activity quantified by the DSS is shown on the right. Data are from screens with CD28 costimulation. Enlarged points indicate drugs with T-cell reporter DSS >15 or CAR T-cell cytotoxicity DSS below −25, with selected drugs labeled, and drugs with high inhibition of both cytotoxicity and TCR signaling circled. (B) Dose-response curves of NFAT, NF-κB, and AP-1 reporter activity for selected drugs. Data are from screens with CD28 costimulation. (C) Dose-response curves of CAR T-cell cytotoxicity for selected drugs. (D) Schematic of proposed mechanisms of CAR T-cell cytotoxicity and TCR signaling inhibition by selected drugs of 50 most potent CAR T-cell cytotoxicity inhibitors. Average DSSs of AP-1 (blue), NF-kB (green), and NFAT (red) inhibition for the listed drugs in the CD28 costimulation screens are shown as bar plots.
Comparison of the DSS reflecting inhibition of TCR signaling and CAR T-cell cytotoxicity identified several compounds potently inhibiting both functions (Figure 2A). The calcineurin inhibitor tacrolimus, MAPK pathway inhibitors refametinib and trametinib, and TKIs targeting SRC/LCK (ponatinib, dasatinib, and saracatinib) strongly suppressed TCR signaling, implying that these compounds impair cytotoxicity largely by inhibiting CAR signaling (Figure 2A-C). In contrast, JAK and PI3K inhibitors had negligible effects on TCR signaling, yet they potently suppressed cytotoxicity, suggesting other mechanisms of action for JAK inhibitors and potentially reflecting constitutive PI3K signaling in Jurkat cells in the case of PI3K inhibitors.34 Several drugs preferentially inhibited certain transcription factors, such as tacrolimus and SRC inhibitors, which strongly inhibited NFAT, and MAPK inhibitors, which targeted both NFAT and AP-1 (Figure 2D). Of note, most drug responses were similar with or without CD28 costimulation, suggesting that the inhibitory effects are not specific to CD28-mediated costimulation and may be generalizable to CARs containing different costimulatory domains (supplemental Figure 2D). Importantly, SMAC mimetics identified as enhancers of CAR T-cell cytotoxicity did not substantially influence TCR signaling, suggesting a different mechanism of action (supplemental Figure 3).
Validation of top candidate drugs influencing CAR T-cell cytotoxicity across B-cell malignancies
To validate the results from the NALM6-luc screen, we tested 31 highly sensitizing and inhibitory compounds, as well as drugs clinically used in B-cell malignancies (eg, ibrutinib and dexamethasone) using CAR T cells from different donors and genetically and phenotypically different target cells (Figure 3A). Screens performed with cells from different donors agreed strongly, suggesting robustness of the assay and generalizability of the observed drug effects to different donors (Figure 3B). Importantly, validation screens performed with control T cells without the CD19 CAR showed similarity to screens using only cancer cells, indicating that the observed effects are specific to CAR T cells activated by CD19 engagement (supplemental Figure 4).
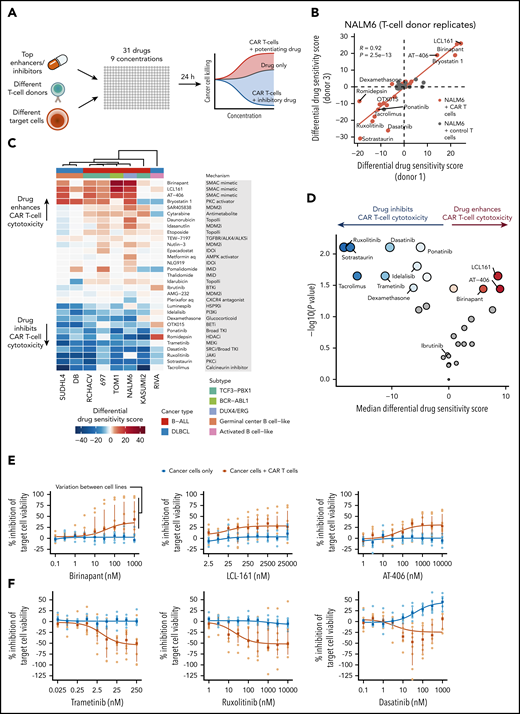
Validation of top candidate drugs influencing CAR T-cell cytotoxicity across B-cell malignancies. (A) Schematic of validation experiments performed using different T-cell donors and target cell lines with a focused 31-drug panel spanning 9 drug concentrations. (B) Comparison of differential DSSs (CAR T-cell or control T-cell coculture compared with NALM6 alone) between experiments performed using CAR T cells from different donors. Correlation coefficient R and P values are calculated using Pearson correlation of the NALM6 plus CAR T-cell coculture scores shown in orange. (C) Heatmap of differential DSSs (CAR T-cell coculture compared with target cells alone) across 8 cell lines representing B-ALL and DLBCL. Drugs are ordered by average differential DSS, and cell lines are clustered using Spearman correlation distance and Ward’s linkage. (D) Volcano plot of drug response differences between validation drug screens with CAR T-cell exposure or with target cells alone. Vertical axis and dot size show the significance of DSS difference between CAR T cell–treated and target-only conditions for each drug as negative log10 of the P value obtained using Wilcoxon signed-rank test. Horizontal axis and the blue and red color intensities show the median differential DSS for each drug. Drugs with FDR >10% (Benjamini-Hochberg procedure) are colored gray. (E) Dose-response curves of CAR T-cell cytotoxicity for the SMAC mimetics birinapant, LCL-161, and AT-406 across the 8 cell lines. Orange curve and points indicate drug responses with coculture of CAR T cells and target cells, and blue curve and points indicate drug responses with target cells only. Curves are drawn using the median percent inhibition values across cell lines shown as rectangles; dots indicate individual cell line percent inhibition values; vertical lines indicate interquartile ranges. (F) Dose-response curves of CAR T-cell cytotoxicity for selected inhibitory drugs across the 8 cell lines as in panel E.
Validation of top candidate drugs influencing CAR T-cell cytotoxicity across B-cell malignancies. (A) Schematic of validation experiments performed using different T-cell donors and target cell lines with a focused 31-drug panel spanning 9 drug concentrations. (B) Comparison of differential DSSs (CAR T-cell or control T-cell coculture compared with NALM6 alone) between experiments performed using CAR T cells from different donors. Correlation coefficient R and P values are calculated using Pearson correlation of the NALM6 plus CAR T-cell coculture scores shown in orange. (C) Heatmap of differential DSSs (CAR T-cell coculture compared with target cells alone) across 8 cell lines representing B-ALL and DLBCL. Drugs are ordered by average differential DSS, and cell lines are clustered using Spearman correlation distance and Ward’s linkage. (D) Volcano plot of drug response differences between validation drug screens with CAR T-cell exposure or with target cells alone. Vertical axis and dot size show the significance of DSS difference between CAR T cell–treated and target-only conditions for each drug as negative log10 of the P value obtained using Wilcoxon signed-rank test. Horizontal axis and the blue and red color intensities show the median differential DSS for each drug. Drugs with FDR >10% (Benjamini-Hochberg procedure) are colored gray. (E) Dose-response curves of CAR T-cell cytotoxicity for the SMAC mimetics birinapant, LCL-161, and AT-406 across the 8 cell lines. Orange curve and points indicate drug responses with coculture of CAR T cells and target cells, and blue curve and points indicate drug responses with target cells only. Curves are drawn using the median percent inhibition values across cell lines shown as rectangles; dots indicate individual cell line percent inhibition values; vertical lines indicate interquartile ranges. (F) Dose-response curves of CAR T-cell cytotoxicity for selected inhibitory drugs across the 8 cell lines as in panel E.
We generated luciferase-expressing cell lines from a panel of CD19+ B-cell malignancies, including the B-ALL cells 697, KASUMI2, RCHACV, and TOM1 and the DLBCL cell lines DB, RIVA, and SUDHL4, and performed validation screens (supplemental Table 4). SMAC mimetics were able to potentiate CAR T-cell cytotoxicity, especially in NALM6 and TOM1 and to a slightly lesser extent in RCHACV, SUDHL4, and 697, whereas DB, RIVA, and KASUMI2 showed no or very limited responses (Figure 3C-E). In addition to SMAC mimetics, we observed potentiating effects with the PKC activator bryostatin-1. The inhibitory effects of MEK, JAK, SRC, and calcineurin inhibitors were largely uniform across target cell lines, consistent with mechanisms relying primarily on inhibition of T-cell function (Figure 3F). In contrast, dexamethasone only minimally affected CAR T-cell cytotoxicity, and no effect was observed with ibrutinib (Figure 3D).
SMAC mimetics sensitize primary B-ALL cells to CAR T-cell cytotoxicity
We next evaluated whether SMAC mimetics could sensitize primary B-ALL cells to CAR T-cell lysis ex vivo using a flow cytometry–based cytotoxicity assay (Figure 4A; supplemental Figure 5A). Treatment with birinapant was able to augment CAR T-cell cytotoxicity against primary leukemia cells, although heterogeneity across patients similarly to the cell lines was evident (Figure 4B). The additional decrease in target cell viability achieved by birinapant treatment was evident at various effector/target cell ratios and drug concentrations (supplemental Figure 5B-D). Of note, birinapant did not influence CD19 expression, which was high in all tested B-ALL patients, indicating that drug-induced alterations of target antigen expression do not explain the sensitization (supplemental Figure 5E-F). Interestingly, Ph+ and Ph-like ALL cells were more sensitive to the combination (Figure 4C-D), consistent with the high sensitivity of the Ph+ ALL cell line TOM1.
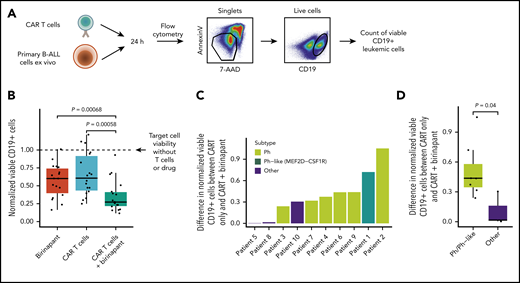
SMAC mimetics sensitize primary B-ALL cells to CAR T-cell cytotoxicity. (A) Schematic of flow cytometry–based drug sensitivity profiling of primary B-ALL sensitivity to CAR T cells ex vivo. (B) Drug sensitivity profiling of primary B-ALL sensitivity to CAR T cells (1:8 effector/target ratio) with and without the SMAC mimetic birinapant ex vivo. CD19+ target cell viability is normalized to the untreated condition without CAR T cells and with DMSO. P values from 2-tailed Wilcoxon signed-rank test are shown. (C) Differences in normalized viable CD19+ primary leukemic cells between cells treated with CAR T cells at 1:8 effector/target ratio and combination of CAR T cells with birinapant. Higher value indicates higher cell death achieved by the combination treatment compared with CAR T cells only. The patient samples are ranked by increasing difference in viability, and bars are colored by genetic subtype of the leukemic cells. (D) Differences in normalized viable CD19+ leukemic cells between cells treated with CAR T cells and combination of CAR T cells with birinapant compared between Philadelphia chromosome (Ph)/Ph-like ALLs (n = 7) and other (Ph−/non–Ph-like) genetic subtypes (n = 3). P value from 2-tailed Wilcoxon rank sum test is shown.
SMAC mimetics sensitize primary B-ALL cells to CAR T-cell cytotoxicity. (A) Schematic of flow cytometry–based drug sensitivity profiling of primary B-ALL sensitivity to CAR T cells ex vivo. (B) Drug sensitivity profiling of primary B-ALL sensitivity to CAR T cells (1:8 effector/target ratio) with and without the SMAC mimetic birinapant ex vivo. CD19+ target cell viability is normalized to the untreated condition without CAR T cells and with DMSO. P values from 2-tailed Wilcoxon signed-rank test are shown. (C) Differences in normalized viable CD19+ primary leukemic cells between cells treated with CAR T cells at 1:8 effector/target ratio and combination of CAR T cells with birinapant. Higher value indicates higher cell death achieved by the combination treatment compared with CAR T cells only. The patient samples are ranked by increasing difference in viability, and bars are colored by genetic subtype of the leukemic cells. (D) Differences in normalized viable CD19+ leukemic cells between cells treated with CAR T cells and combination of CAR T cells with birinapant compared between Philadelphia chromosome (Ph)/Ph-like ALLs (n = 7) and other (Ph−/non–Ph-like) genetic subtypes (n = 3). P value from 2-tailed Wilcoxon rank sum test is shown.
Given reports on both stimulatory and inhibitory effects of IAP inhibition on T cells,35,36 we studied the long-term effects of the SMAC mimetics on CAR T-cell ex vivo expansion and phenotype (supplemental Figure 6). CAR T cells expanded >100-fold on days 10 to 22 after CAR T-cell generation, despite a slight decrease in expansion rate upon treatment with birinapant or LCL-161, suggesting lack of overt toxicity of IAP inhibition on CAR T-cell expansion. In addition, the progression of the CAR T-cell phenotype toward effector T cells during expansion was largely unaltered by SMAC mimetics.
CRISPR screens identify death receptor signaling as essential for CAR T-cell cytotoxicity and SMAC mimetic function
Next, to identify cancer cell–intrinsic factors essential for CAR T-cell cytotoxicity using a complementary genetic approach, we conducted genome-wide CRISPR screens. We infected Cas9-expressing NALM6 with a genome-scale lentiviral sgRNA library and exposed the cell pool to CAR T cells (Figure 5A). After 9 to 12 days of culture, genomic DNA was sequenced to determine the relative enrichment of sgRNAs in CAR T cell-treated vs untreated conditions. Disruption of genes enriched in CAR T cell–treated NALM6 leads to decreased sensitivity to CAR T-cell cytotoxicity, indicating essentiality of these genes for CAR T-cell cytotoxicity. To integrate the CRISPR-based functional genomics with results from the drug profiling and shed light on the mechanism by which SMAC mimetics synergize with CAR T cells, we included a condition with SMAC mimetic plus CAR T-cell combination.
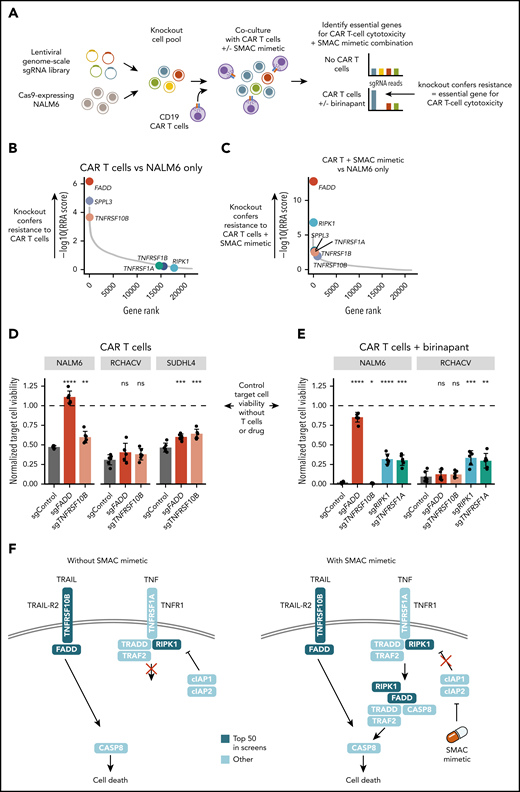
Genome-wide CRISPR screens identify essential genes for CAR T-cell cytotoxicity and SMAC mimetic function. (A) Schematic of the CRISPR-Cas9 screening approach. (B) Positively selected hits of NALM6 CRISPR screen with CAR T-cell coculture compared with NALM6 only. Genes are ranked by negative log10 of the MAGeCK RRA score for positive selection. (C) Positively selected hits of NALM6 CRISPR screen with CAR T cells plus SMAC mimetic compared with NALM6 only. Genes are ranked by negative log10 of MAGeCK RRA score for positive selection. (D) Validation of the effects of FADD and TNFRSF10B disruption in the B-ALL cell lines NALM6 and RCHACV and the DLBCL cell line SUDHL4 exposed to CAR T cells alone. Bar plots show viability of target cells expressing FADD, TNFRSF10B, or control sgRNAs exposed to CAR T cells for 24 hours (1:4 effector/target ratio) treated with DMSO. (E) Validation of the effects of FADD, TNFRSF10B, RIPK1, and TNFRSF1A disruption in NALM6 and RCHACV exposed to CAR T cells and SMAC mimetic. Bar plots show viability of target cells expressing FADD, TNFRSF10B, RIPK1, TNFRSF1A, or control sgRNAs exposed to CAR T cells for 24 hours (1:4 effector/target ratio) treated with birinapant. In panels D and E, the data are normalized to DMSO-treated target cells without T cells (indicated as dashed line). Bar heights represent average of 6 technical replicates for each sgRNA, dots represent technical replicates, and error bars show standard deviation. P values are calculated using Welch’s t test between the indicated condition and single-guide control. (F) Schematic of proposed mechanism of CAR T-cell cytotoxicity and SMAC mimetic–mediated sensitization to CAR T-cell death receptor–mediated cytotoxicity. Genes within top 50 hits in CRISPR screens are colored in dark blue. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant (P ≥ .05).
Genome-wide CRISPR screens identify essential genes for CAR T-cell cytotoxicity and SMAC mimetic function. (A) Schematic of the CRISPR-Cas9 screening approach. (B) Positively selected hits of NALM6 CRISPR screen with CAR T-cell coculture compared with NALM6 only. Genes are ranked by negative log10 of the MAGeCK RRA score for positive selection. (C) Positively selected hits of NALM6 CRISPR screen with CAR T cells plus SMAC mimetic compared with NALM6 only. Genes are ranked by negative log10 of MAGeCK RRA score for positive selection. (D) Validation of the effects of FADD and TNFRSF10B disruption in the B-ALL cell lines NALM6 and RCHACV and the DLBCL cell line SUDHL4 exposed to CAR T cells alone. Bar plots show viability of target cells expressing FADD, TNFRSF10B, or control sgRNAs exposed to CAR T cells for 24 hours (1:4 effector/target ratio) treated with DMSO. (E) Validation of the effects of FADD, TNFRSF10B, RIPK1, and TNFRSF1A disruption in NALM6 and RCHACV exposed to CAR T cells and SMAC mimetic. Bar plots show viability of target cells expressing FADD, TNFRSF10B, RIPK1, TNFRSF1A, or control sgRNAs exposed to CAR T cells for 24 hours (1:4 effector/target ratio) treated with birinapant. In panels D and E, the data are normalized to DMSO-treated target cells without T cells (indicated as dashed line). Bar heights represent average of 6 technical replicates for each sgRNA, dots represent technical replicates, and error bars show standard deviation. P values are calculated using Welch’s t test between the indicated condition and single-guide control. (F) Schematic of proposed mechanism of CAR T-cell cytotoxicity and SMAC mimetic–mediated sensitization to CAR T-cell death receptor–mediated cytotoxicity. Genes within top 50 hits in CRISPR screens are colored in dark blue. *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant (P ≥ .05).
MAGeCK-based data analysis29 identified FADD as the most enriched gene in CAR T cell–treated NALM6 compared with the control (Figure 5B; supplemental Figure 7; supplemental Table 5). FADD encodes a mediator involved in death receptor–mediated apoptosis.37,38 Together with enrichment of TNFRSF10B encoding the TRAIL-R2 death receptor (Figure 5B), the CRISPR screen data indicate an essential role of death receptor–mediated apoptosis for effective CAR T-cell cytotoxicity. In the CAR T-cell plus SMAC mimetic resistance screen, FADD again emerged as the most enriched gene, followed by RIPK1, disruption of which had no effect in NALM6 treated with only CAR T cells (Figure 5C), suggesting even further sensitization to death receptor apoptosis involving FADD upon SMAC mimetic treatment. RIPK1 regulates the formation of complex 2 required for TNF-mediated apoptosis and is itself regulated by IAPs targeting RIPK1 to degradation through ubiquitination.38 Consistently, components of the TNF receptor complex (TNFRSF1A and TNFRSF1B) ranked higher in the combination screen compared with the screen with CAR T cells only (Figure 5B-C). The screens also identified genes not previously associated with T cell–mediated cytotoxicity, such as SPPL3, disruption of which conferred resistance to CAR T-cell cytotoxicity (supplemental Figure 8A-B), and mir-4661, silencing of which further sensitized to the CAR T-cell plus SMAC mimetic combination (supplemental Figure 7B,D).
Heterogeneity in death receptor dependence of CAR T-cell cytotoxicity in B-cell malignancies
Validation experiments using NALM6 expressing Cas9 and individual sgRNAs targeting screen hits revealed complete abrogation of CAR T cell–mediated cytotoxicity in cells expressing FADD sgRNAs (Figure 5D; supplemental Figure 8). TNFRSF10B disruption conferred resistance to CAR T cells to a lesser extent compared with FADD, suggesting that other death receptors acting through FADD also mediate CAR T-cell cytotoxicity against NALM6.
We next tested whether the discovered genetic mechanisms could be generalized beyond the NALM6 model. Interestingly, disruption of FADD or TNFRSF10B in the B-ALL cell line RCHACV did not confer resistance to CAR T cells (Figure 5D). In contrast, the DLBCL cell line SUDHL4 showed results consistent with those obtained in NALM6, suggesting that death receptor signaling can mediate CAR T-cell cytotoxicity in B-cell lymphomas as well. The higher surface expression of TRAIL-R2 (TNFRSF10B) in NALM6 and SUDHL4 compared with RCHACV may explain the observed differences. Together, the data indicate that the dependence of CAR T-cell cytotoxicity on death receptor signaling is heterogeneous among individual cancers.
In NALM6, RIPK1 or TNFRSF1A disruption did not alter sensitivity to CAR T cells alone, but it rescued NALM6 cells from SMAC mimetic–induced sensitization, confirming the TNF-dependent mechanism (Figure 5E). Across different effector/target ratios, sgRNAs targeting RIPK1 consistently restored the viability of CAR T-cell plus birinapant–treated NALM6 to levels similar to control NALM6 treated with CAR T cells alone, indicating that RIPK1 is required for the SMAC mimetic–induced sensitization without influencing sensitivity to CAR T cells alone (supplemental Figure 8C). Consistently with NALM6, sgRNAs targeting RIPK1 or TNFRSF1A rescued RCHACV cells from SMAC mimetic–induced sensitization (Figure 5E). Although TNFRSF1A was required for SMAC mimetic function, the surface expression of TNFRSF1A did not explain differences in sensitivity across the cell lines (supplemental Figure 10).
To investigate how cancer cells resistant to FADD-mediated cytotoxicity influence CAR T cells, we examined CAR T-cell proliferation upon exposure to cells carrying FADD sgRNAs (supplemental Figure 11). Coculture with these modified target cells did not drastically alter the CAR T-cell proliferation response compared with control NALM6 in a short-term assay (4 days), although we noted that target cells carrying FADD sgRNAs triggered slightly more extensive CAR T-cell proliferation in a long-term (2 weeks) assay.
Together, the data point toward a model where death receptor signaling is essential for CAR T-cell cytotoxicity to a varying extent across cancers, with further sensitization and involvement of RIPK1/TNF-mediated death receptor signaling upon inhibition of IAPs by SMAC mimetics (Figure 5F).
Death receptor expression varies across genetic subtypes of B-cell malignancies
Analysis of genomic data sets of various B-cell malignancies32,39 revealed heterogeneity in death receptor gene expression (supplemental Table 6; supplemental Figure 12). The TRAIL receptors TNFRSF10B and TNFSRF10A were highly expressed in chronic lymphocytic leukemia compared with other cancers (hypergeometric test, FDR < 10−45; Figure 6A). Within B-ALL,40 TNFRSF10B expression was particularly low in subtypes harboring MEF2D (Wilcoxon rank sum test, FDR = 6.4 × 10−21) or TCF3-PBX1 fusions (FDR = 2.1 × 10−18), in contrast to Ph+ B-ALL characterized by high death receptor expression (Figure 6B). These data are consistent with the high sensitivity of primary Ph+ B-ALL cells and TOM1 to CAR T-cell plus SMAC mimetic treatment, as opposed to the lower sensitivity observed in 697, KASUMI2, and RCHACV harboring TCF3-PBX1 (Figure 3C). We found no difference in TNFRSF10B expression between high- and standard-risk B-ALL (supplemental Figure 12D). In DLBCL, TNFRSF10B expression correlated negatively with TP53 mutations, consistent in 2 independent data sets41 (Figure 6C). Thus, heterogeneity in death receptor expression linked to cancer genetics may affect sensitivity to death receptor–mediated cytotoxicity of CAR T cells.
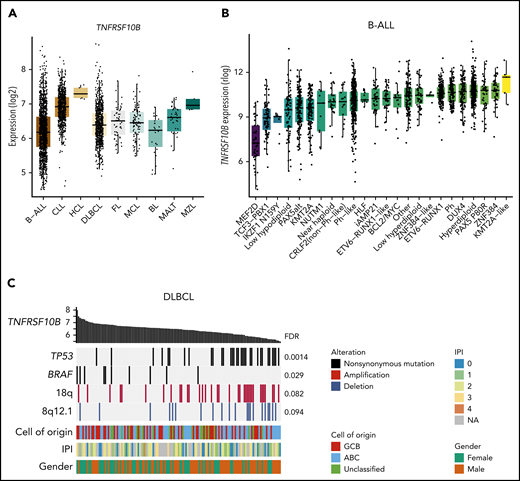
Death receptor expression varies across genetic subtypes of B-cell malignancies. (A) Expression of TNFRSF10B across B-cell malignancies in data from the Hemap resource containing aggregated transcriptomic profiles across hematological malignancies. (B) Expression of TNFRSF10B across B-ALL subtypes in data by Gu et al.40 (C) Genetic alterations most significantly associated with TNFRSF10B expression (log2 expression shown as bar plot) in Chapuy et al.41 DLBCL data shown as an oncoprint. FDR for the significance of Spearman correlations between TNFRSF10B expression and alterations are shown. ABC, activated B cell; BL, Burkitt lymphoma; CLL, chronic lymphocytic leukemia; FL, follicular lymphoma; GCB, germinal center B cell; HCL, hairy cell leukemia; IPI, International Prognostic Index; MALT, mucosa-associated lymphoid tissue lymphoma; MCL, mantle cell lymphoma; MZL, marginal zone lymphoma.
Death receptor expression varies across genetic subtypes of B-cell malignancies. (A) Expression of TNFRSF10B across B-cell malignancies in data from the Hemap resource containing aggregated transcriptomic profiles across hematological malignancies. (B) Expression of TNFRSF10B across B-ALL subtypes in data by Gu et al.40 (C) Genetic alterations most significantly associated with TNFRSF10B expression (log2 expression shown as bar plot) in Chapuy et al.41 DLBCL data shown as an oncoprint. FDR for the significance of Spearman correlations between TNFRSF10B expression and alterations are shown. ABC, activated B cell; BL, Burkitt lymphoma; CLL, chronic lymphocytic leukemia; FL, follicular lymphoma; GCB, germinal center B cell; HCL, hairy cell leukemia; IPI, International Prognostic Index; MALT, mucosa-associated lymphoid tissue lymphoma; MCL, mantle cell lymphoma; MZL, marginal zone lymphoma.
Discussion
The influence of cancer drugs on T-cell effector function and the mechanisms of CAR T-cell cytotoxicity are incompletely understood. We profiled the pharmacological effects of >500 drugs on multiple aspects of T-cell function, including cytotoxicity and TF activity downstream of TCR signaling, and investigated genes the loss of which in cancer cells impaired the effector function of CAR T cells using genome-scale CRISPR screens. The integration of the complementary drug profiling and CRISPR-based functional genomics allowed systematic identification of the mechanism of action of compounds highlighted in the small-molecule screens.
We identified SMAC mimetics as potent sensitizers of CD19+ B-cell malignancies to CAR T-cell cytotoxicity. SMAC mimetics have been suggested as immunotherapy combinations in solid tumors42-44 and multiple myeloma45 through several distinct mechanisms and could therefore enhance cancer immunotherapies by targeting multiple arms of the antitumor immune response. In the clinical setting, SMAC mimetics could potentially be used in cases refractory to CAR T-cell therapy. Furthermore, pharmacological sensitization to CAR T-cell cytotoxicity could enable the use of reduced doses of CAR T cells, potentially leading to fewer toxicities.46 The potential of SMAC mimetics to broaden the spectrum of cancer types amenable to CAR T-cell therapy is an important avenue for further investigation, and data from a HER2-transgenic mouse colorectal cancer model supports the use of these drugs in combination with CAR T cells.44 The concentration of SMAC mimetics required for the CAR T-cell sensitizing effect is within the range of clinically achievable plasma concentrations,47,48 suggesting potential for clinical translation. The SMAC mimetic birinapant is currently being investigated in combination with PD-1 blockade immunotherapy (registered at www.clinicaltrials.gov as #NCT02587962), and results from this study should provide valuable information on the feasibility, dosing regimen, and safety profile of a potential combination with CAR T-cell immunotherapy. However, safety issues such as the potential impact of the cytotoxicity-sensitizing effect of SMAC mimetics on healthy tissues, risk for cytokine release syndrome reported with both CAR T-cell2,46 and SMAC mimetic treatments,48,49 and influence of SMAC mimetics on long-term persistence of CAR T cells in vivo should be examined before clinical translation.
TKIs targeting the MAPK pathway, SRC, PI3K, and JAK compromised the cytotoxic function of CAR T cells in our preclinical assays, warranting careful consideration when combining these drugs with T-cell immunotherapies for example when treating Ph+ or Ph-like B-ALL. The drugs highlighted here as potent suppressors of cytotoxicity and T-cell activation may also provide candidates for new treatment approaches to alleviating immune-mediated adverse effects of immunotherapies, such as CAR T-cell therapy–related toxicities or T cell–related autoimmune disorders. Consistent with our results, the SRC inhibitor dasatinib, identified as a suppressor of both CAR T-cell cytotoxicity and TCR signaling in our screens, was recently suggested as a pharmacological on/off switch to control CAR T cell–induced toxicities.50,51 Furthermore, JAK inhibitors are established treatment options in many immune and inflammatory conditions.52 In contrast, dexamethasone, clinically used to treat CAR T-cell therapy–related toxicities, only minimally affected CAR T-cell cytotoxicity, suggesting that dexamethasone may alleviate toxicities without compromising the antileukemia function of CAR T cells, as indicated by clinical experience as well.53 We observed a similar lack of interference with CAR T-cell cytotoxicity for ibrutinib, investigated in combination with CAR T cells.54
T-cell cytotoxicity can occur through the release of cytolytic granule contents or alternatively through death receptor ligands including FAS ligand, TRAIL, and TNF.13 In the CAR T-cell setting, our CRISPR screen data highlight the essential role of death receptor–mediated cytotoxicity through FADD and TNFRSF10B (TRAIL-R2). In line with observations by us and others55 in B-cell malignancies, CAR T cells were recently shown to induce TRAIL expression upon target engagement and mediate TRAIL-dependent cytotoxicity after radiation preconditioning in a model of pancreatic adenocarcinoma.56 Importantly, we found heterogeneity among cancers regarding the relative importance of death receptor signaling to effective CAR T-cell cytotoxicity. The differential expression of death receptors across subtypes of B-cell malignancies may thus influence sensitivity to CAR T-cell cytotoxicity and explain primary and adaptive resistance to CAR therapies.
By combining genome-wide genetic perturbation screening with drug profiling, we dissected the mechanism of action of a complex pharmacological effect involving the interaction of 2 distinct cell types. The CRISPR screen data highlight that inhibition of IAPs by SMAC mimetics results in further sensitization to death receptor–mediated CAR T-cell cytotoxicity through an RIPK1-mediated mechanism. The dependence of SMAC mimetic–induced sensitization in RCHACV cells on RIPK1 and TNFRSF1A but not on FADD may indicate involvement of other types of cell death, such as necroptosis, in contrast to FADD-mediated apoptosis.57 Importantly, the abrogation of the sensitizing effect by loss of FADD or RIPK1 in the cancer cells pinpoints the mechanism of action of SMAC mimetics to the cancer cells, as opposed to potentiating effects on T cells. Consistently, SMAC mimetics did not show significant effects in TCR reporter assays, whereas these assays suggested the effect of some drugs to occur mostly on the T-cell side, such as the inhibitory effects of MAPK and SRC inhibitors. By profiling several target cell lines and primary cultures, we uncovered heterogeneity in the interactions of T cells and small molecules, potentially related to the genetic makeup of the cancer cells. The differential susceptibility of distinct leukemia subtypes to further sensitization by SMAC mimetics indicates a more general rationale for tailoring small-molecule drug combinations with immunotherapy based on genetic and phenotypic properties of cancer cells.
The wide concentration range used in our drug profiling assays allowed accurate assessment of the dose-response properties of the immunomodulatory effects both on cytotoxicity and TCR signaling TF activity, providing a resource for further research into drug-induced immunomodulation. Although the differential DSS metric has been used for systematic analysis of drug responses, detailed dose-response data at each concentration are provided for easier evaluation of the magnitude of the drug responses and for guiding further experimentation (supplemental Tables 1-4). Limitations of our in vitro screening approaches include the inability to assess the influence of drugs or genes in different microenvironments encountered in vivo or long-term effects such as the phenotype, exhaustion, and persistence of CAR T cells. We envision that our high-throughput screens focusing on the cytotoxic effector function of CAR T cells can provide a starting point for specific studies on individual compounds or genes. Moreover, although the TCR signaling reporter assay system has limitations resulting from genetic and functional aberrations in the leukemic Jurkat cells,34,58 we believe that our screening data provide a valuable resource on the effects of oncology drugs in this model system that has provided much of the current knowledge on TCR signaling.59-62 Finally, CARs harboring different costimulatory domains such as 4-1BB may behave differently with respect to drug sensitivity,63-65 and only effects on CAR T cells targeting CD19 were tested.
In summary, our pharmacological and genetic screens investigating CAR T-cell cytotoxicity converged on death receptor signaling, suggesting that death receptor–mediated cytotoxicity may be an important mechanism of cancer cell elimination by CAR T cells. The drug profiling data provide a framework to understand the immunomodulatory properties of cancer drugs regarding CAR T-cell cytotoxicity and TCR signaling.
High-throughput drug profiling and CRISPR screen data are provided as supplementary data.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Laura Turunen, Jani Saarela, Katja Suomi, and Maria Nurmi of the High-Throughput Biomedicine Unit and the personnel of the Sequencing Laboratory at the Institute of Molecular Medicine Finland. The authors also thank Caroline Heckman, Sirpa Leppä, and Olli Lohi for collaboration by providing the cell lines used in the study. The Biomedicum Helsinki Flow Cytometry Core Unit and the Biomedicum Virus Core are acknowledged for their services. The primary B-ALL samples were provided by the Finnish Hematology Registry and Clinical Biobank (FHRB). We thank all the patients for their generous participation. The FHRB biobank is supported by the Finnish Association of Hematology, Finnish Red Cross Blood Service, Institute for Molecular Medicine Finland, and participating hospitals in Finland.
This study was supported by the Cancer Foundation Finland, the Sigrid Juselius Foundation, the Relander Foundation, state funding for university-level health research in Finland, and HiLife fellow funds from the University of Helsinki. O.D. was supported by grants from the Biomedicum Helsinki Foundation and the Finnish Medical Society. J. Koski was supported by grants from the Finnish Pediatric Cancer Foundation, the Väre Pediatric Research Foundation, the Cancer Foundation Finland, the Orion Research Foundation, and state funding for university-level health research in Finland.
Authorship
Contribution: O.D. conceived and designed the study, performed the drug profiling experiments, CRISPR screens, and validation experiments, analyzed the data, and wrote the paper; J. Koski and P.M. performed CAR T-cell generation and expansion; J. Koski designed the luciferase-GFP construct, generated luciferase-expressing cell lines, and performed validation experiments; A.I. analyzed drug profiling data; J. Klievink generated the NALM6-luc and NALM6-Cas9 cell lines and cell lines for individual sgRNA validations and performed molecular cloning, lentivirus production, and CRISPR screens; J.L. generated TCR signaling reporter cell lines; P.P. analyzed gene expression and genomic data sets; H.H. performed primary B-ALL cultures; K.S. participated in performing CRISPR screens; T.H. performed sequencing of CRISPR screen samples; P.E. participated in CRISPR screen design; P.S. provided TCR signaling reporter cell lines and participated in assay design; M. Kankainen participated in CRISPR screen design and data analysis; T.A. designed and supervised drug profiling data analyses; M.A.I.K., M. Korhonen, and S.M. supervised the study and wrote the paper; and all authors contributed to writing the paper and approved the final version.
Conflict-of-interest disclosure: Labcyte, Inc., and FIMM/University of Helsinki have a collaboration agreement on the utilization of Labcyte’s acoustic dispensing technologies. S.M. has received honoraria and research funding from Novartis, Pfizer, and Bristol-Myers Squibb (not related to this study). H.H. has received research funding from Incyte. The remaining authors declare no competing financial interests.
Correspondence: Satu Mustjoki, University of Helsinki, Haartmaninkatu 8, POB 63, 00014 Helsinki, Finland; e-mail: satu.mustjoki@helsinki.fi; and Olli Dufva, University of Helsinki, Haartmaninkatu 8, POB 63, 00014 Helsinki, Finland; e-mail: olli.dufva@helsinki.fi.