Key Points
AGK deficiency reduces thrombopoiesis, and AGK regulates megakaryocyte differentiation by maintaining JAK2/Stat3 signaling activation.
Peptide 617F augments JAK2/Stat3 signaling by promoting the association of AGK and JAK2 and accelerates megakaryocyte differentiation.

Abstract
Abnormal megakaryocyte development and platelet production lead to thrombocytopenia or thrombocythemia and increase the risk of hemorrhage or thrombosis. Acylglycerol kinase (AGK) is a mitochondrial membrane kinase that catalyzes the formation of phosphatidic acid and lysophosphatidic acid. Mutation of AGK has been described as the major cause of Sengers syndrome, and the patients with Sengers syndrome have been reported to exhibit thrombocytopenia. In this study, we found that megakaryocyte/platelet-specific AGK-deficient mice developed thrombocytopenia and splenomegaly, mainly caused by inefficient bone marrow thrombocytopoiesis and excessive extramedullary hematopoiesis, but not by apoptosis of circulating platelets. It has been reported that the G126E mutation arrests the kinase activity of AGK. The AGK G126E mutation did not affect peripheral platelet counts or megakaryocyte differentiation, suggesting that the involvement of AGK in megakaryocyte development and platelet biogenesis was not dependent on its kinase activity. The Mpl/Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (Stat3) pathway is the major signaling pathway regulating megakaryocyte development. Our study confirmed that AGK can bind to JAK2 in megakaryocytes/platelets. More interestingly, we found that the JAK2 V617F mutation dramatically enhanced the binding of AGK to JAK2 and greatly facilitated JAK2/Stat3 signaling in megakaryocytes/platelets in response to thrombopoietin. We also found that the JAK2 JAK homology 2 domain peptide YGVCF617CGDENI enhanced the binding of AGK to JAK2 and that cell-permeable peptides containing YGVCF617CGDENI sequences accelerated proplatelet formation. Therefore, our study reveals critical roles of AGK in megakaryocyte differentiation and platelet biogenesis and suggests that targeting the interaction between AGK and JAK2 may be a novel strategy for the treatment of thrombocytopenia or thrombocythemia.
Introduction
Platelets are essential components of the circulating hematologic system and play vital roles in thrombus formation, hemostasis, inflammation, and tumor metastasis.1-3 Megakaryocytes are responsible for the production of platelets in the hierarchical hematopoietic system. Innate promegakaryocytes derived from hematopoietic stem cells (HSCs) undergo a series of complex biological processes, such as endomitosis, polyploid formation, megakaryocyte demarcation membrane system maturation, dense tubular system maturation, and secretory granule formation, to form mature megakaryocytes.4-8
Impaired proliferation or maturation of megakaryocytes in bone marrow (BM) is a major cause of thrombocytopenia, which increases the risk of vascular, mucosal, and visceral hemorrhage.9-11 In addition, hyperactive megakaryocyte development is observed in myeloproliferative neoplasms (MPNs) and gives rise to thrombocythemia. Patients with thrombocythemia have elevated risks of arterial thrombosis and venous thrombosis.12-14
The thrombopoietin (TPO)/Mpl/Janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (Stat3) signaling pathway is the major pathway regulating megakaryocyte development and platelet formation. TPO is produced mostly in hepatocytes. TPO binds and signals through the Mpl receptor in megakaryocytes and HSCs, leading to dimerization of the Mpl receptors and phosphorylation of JAK2. Tyr626 and Tyr631 in Mpl are then in turn phosphorylated by JAK2, which recruits signaling molecules containing the Src homology (SH2) domain, such as Stat family molecules.15,16
The JAK homology 2 (JH2) domain in JAK2 is an autoinhibitory domain that suppresses the kinase activity of the JAK homology 1 (JH1) domain. JAK2 V617F in the JH2 domain is a frequent mutation found in MPN patients.17 It is well accepted that the replacement of phenylalanine (F) with valine (V) eliminates the inhibitory effect of the JH2 domain, causes continuous activation of JAK2/Stat3 signaling, and leads to the occurrence of polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF).18,19
Acylglycerol kinase (AGK) was first identified as a mitochondrial inner membrane protein that phosphorylates diacylglycerol and monoacylglycerol to form phosphatidic acid and lysophosphatidic acid.20,21 AGK has also been described as a component of the TIM22 complex independent of kinase activity, and it participates in anchoring processes for some mitochondrial inner membrane proteins.22-24 AGK gene mutations, including start codon mutations, nonsense, frameshift and splice site mutations, cause Sengers syndrome, which is a severe genetic disease characterized by myocardial hypertrophy, congenital cataract, lactic acidosis, and myasthenia.25-27 Among clinical case reports, 2 cases reported the patient with homozygous c.1131+1G>T (p.Ser350Glufs*19) frameshift mutation had thrombocytopenia clinical feature.26,28 However, the role of AGK in thrombopoiesis remains unclear.
In this study, we found that megakaryocyte/platelet-specific AGK-deficient mice developed thrombocytopenia and splenomegaly. We also found that AGK involvement in megakaryocyte differentiation and platelet biogenesis was not dependent on mitochondrial function. A mechanistic study revealed that the JAK2 V617F mutation dramatically enhanced JAK2 binding to AGK and greatly facilitated JAK2/Stat3 signaling in megakaryocytes/platelets in response to TPO. We also found that the JAK2 JH2 domain peptide YGVCF617CGDENI enhanced the binding of AGK to JAK2 and that cell-permeable peptides containing YGVCF617CGDENI sequences accelerated proplatelet formation (PPF). Our study reveals the critical roles of AGK in megakaryocyte differentiation and platelet biogenesis and suggests that targeting the interaction between AGK and JAK2 may be a novel strategy for the treatment of thrombocytopenia or thrombocythemia.
Methods
Antibodies, reagents, and mice
Detailed descriptions of antibodies, reagents, and mice are in supplemental Methods (available on the Blood Web site).
Plasmid construction
Flag-tagged human AGK sequences were PCR amplified from HEK293T cell complementary DNA and cloned into the vector pcDNA3.1-Flag. Hemagglutinin (HA)-tagged human JAK2 sequences were PCR amplified from HEK293T-cell complementary DNA and cloned into the lentiviral vector PLVX-IRES-Puro. AGK G126E and JAK2 V617F mutant plasmid were constructed by using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
Platelet lifespan assay
Endogenous platelet survival was measured after tail intravenous injection of Sulfo-NHS-LC-Biotin (Pierce). Platelets were purified from orbital blood collected at different time points, and the biotin-labeled platelets were detected by flow cytometry with APC Streptavidin (BD Biosciences).
Platelet regeneration
Basal platelet counts were first determined from full blood counts by using a HEMAVET automated hematologic analyzer. Anti-CD42b monoclonal antibodies (2 μg/g weight, Emfret) were then injected into mice IV to deplete platelets. Orbital blood was collected at different time points to monitor platelet regeneration.
Murine splenectomy
Splenectomy was performed as previously described.29 Briefly, mice were anesthetized by intraperitoneal injection with 1% sodium pentobarbital, and their abdomens were shaved and sterilized. A transverse incision 1 cm below the costal margin of the left midabdominal line was made into the abdominal cavity to free the spleen, ligate the splenic artery and vein, and remove the spleen.
Megakaryocyte differentiation, PPF, and polyploid detection
Fetal livers were isolated from 13.5-day-old mouse embryos, homogenized, and cultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum, 1% penicillin/streptomycin stock solution (10 000 U/mL), 10 ng/mL interleukin-3 (IL-3) (Prospec), and 25 IU/mL recombinant human TPO (3SBIO) at 37°C in 5% CO2 for 5 days. Megakaryocytes were then gradient separated with 1.5% bovine serum albumin (BSA) and 3% BSA at 37°C for 1 hour.30
For the megakaryocyte PPF assay, megakaryocytes were cultured on glass slides coated with 50 μg/mL fibrinogen (Sigma) at 37°C. After 12 hours of culture, the megakaryocytes were immobilized with 4% paraformaldehyde and stained with α-tubulin (MilliporeSigma) and Alexa Fluor 488–conjugated anti-mouse immunoglobulin G antibody (Jackson ImmunoResearch Laboratories) for immunofluorescence analysis.
For the megakaryocyte polyploidy assay, megakaryocytes stained with CD41-APC (eBioscience) were fixed overnight at 4°C with 70% ethanol and then incubated for 30 minutes with 50 μg/mL propidium iodide, 100 μg/mL RNase A, and 0.2% Triton X-100. Flow cytometry was then used for analysis of megakaryocyte polyploidy.
Colony-forming assays
Fetal liver cells were cultured for 3 days in the presence of IL-3 and TPO. Lin−Scal-1−c-kit+CD41+CD150+ megakaryocytic progenitors (MKPs) were then sorted by flow cytometry. To determine colony-forming unit megakaryocytes (CFU-MKs), BM cells or sorted fetal liver–derived MKPs were plated on the collagen-based MegaCult medium (StemCell) in the presence of TPO (3SBIO), IL-3 (Prospec), IL-6 (Prospec), and IL-11 (Prospec).31 CFU-MK colonies were evaluated and counted after 7 days according to the manufacturer’s protocol.
Measurement of mitochondrial respiration rate
A Seahorse XFe96 Extracellular Flux Analyzer (Agilent Technologies) was used to measure the mitochondrial respiration rate. Platelets were seeded at a density of 2 × 107 cells per well. For measurement of the oxygen consumption rate (OCR), the platelets were subjected to an XF Cell Mito Stress Test using the following concentrations of injected compounds: 2 μM oligomycin, 0.25 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP), and 1 μM rotenone/antimycin A. The XF Cell Mito Stress Test Kit was purchased from Agilent Technologies.32
Intra-BM injection of peptide
Intra-BM injection was performed as previously described.33 Briefly, 36 hours after injection of anti-CD42b monoclonal antibodies (2 μg/g weight, Emfret) to deplete platelets, mice were anesthetized by intraperitoneal injection with 1% sodium pentobarbital, a pilot hole was drilled through the joint into the right tibia with a 26G needle, and 0.9% NaCl or peptide C-617F (20 μg/g weight; GL Biochem) was injected into Agkf/f and Agk−/− mice.
Statistical analysis
Statistical analyses were implemented with GraphPad Prism 6 (La Jolla, CA). A Student t test was used to determine statistical significance between 2 groups. A value of P ≤ .05 was considered to indicate statistical significance. The statistics can be found in the figure legends.
Results
AGK deficiency caused thrombocytopenia in mice
To explore the functions of AGK in megakaryocyte differentiation, megakaryocyte/platelet-specific AGK-deficient mice (Agkf/fPf4-Cre+, Agk−/−) were constructed. As expected, the protein levels of AGK in platelets from Agk−/− mice were dramatically reduced compared with those in platelets from Agkf/f mice (Figure 1A). Meanwhile, red blood cells (RBCs) and white blood cells (WBC) from Agk−/− mice had the same protein levels of AGK as those from Agkf/f mice (supplemental Figure 1A-B). Then, peripheral blood cell counts were performed. The platelet count results showed that AGK-deficient mice had 619.7 ± 31.44 × 109 circulating platelets/L, whereas control (Agkf/f) mice had 890.6 ± 27.15 × 109 circulated platelets/L (Figure 1B); therefore, AGK deficiency caused thrombocytopenia in mice. Meanwhile, Agkf/f and Agk−/− mice had similar RBC and WBC counts (Figure 1B).
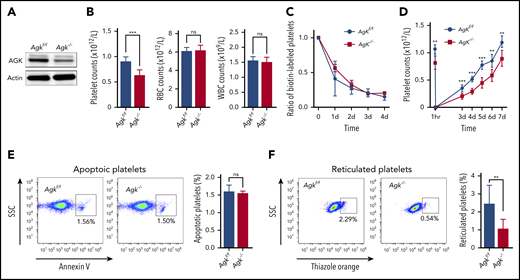
Thrombocytopenia in Agk−/− mice due to impairment of platelet biogenesis. (A) AGK levels were tested in platelets of Agkf/f and Agk−/− mice by western blot analysis. (B) Platelet, RBC, and WBC counts in Agkf/f and Agk−/− mice (n = 12; ***P < .001). (C) Platelet survival was measured after tail IV injection of Sulfo-NHS-LC-Biotin (n = 3). (D) Platelets were first eliminated by tail IV injection of anti-CD42b (2 µg/g), and then platelet counts were monitored with a HEMAVET automated hematologic analyzer at different time points (n = 6; **P < .01, ***P < .001). (E) Platelet apoptosis was measured by annexin V staining using flow cytometry (n = 3). (F) Reticulated platelets were measured by thiazole orange staining using flow cytometry (n = 9; **P < .01). ns, not significant; SSC, side scatter.
Thrombocytopenia in Agk−/− mice due to impairment of platelet biogenesis. (A) AGK levels were tested in platelets of Agkf/f and Agk−/− mice by western blot analysis. (B) Platelet, RBC, and WBC counts in Agkf/f and Agk−/− mice (n = 12; ***P < .001). (C) Platelet survival was measured after tail IV injection of Sulfo-NHS-LC-Biotin (n = 3). (D) Platelets were first eliminated by tail IV injection of anti-CD42b (2 µg/g), and then platelet counts were monitored with a HEMAVET automated hematologic analyzer at different time points (n = 6; **P < .01, ***P < .001). (E) Platelet apoptosis was measured by annexin V staining using flow cytometry (n = 3). (F) Reticulated platelets were measured by thiazole orange staining using flow cytometry (n = 9; **P < .01). ns, not significant; SSC, side scatter.
To explore whether the thrombocytopenia in Agk−/− mice was caused by increased platelet clearance or reduced platelet production, we measured the rates of platelet clearance and regeneration. By measuring the proportions of biotin-labeled platelets at different time points, we determined that Agk−/− mice had normal platelet lifespans (Figure 1C). Anti-CD42b monoclonal antibodies were administered to deplete platelets, and the platelet counts at different time points were measured. The results presented in Figure 1D demonstrate that AGK deficiency significantly impaired thrombopoiesis. Next, the platelets were prepared from peripheral blood (>98% purified platelets are CD41 positive; supplemental Figure 1C) and stained with annexin V and thiazole orange to detect the percentages of apoptotic platelets and reticulated platelets, respectively. The flow cytometry results showed that the percentages of annexin V–positive platelets did not differ between Agk−/− mice (1.55% ± 0.06%) and Agkf/f mice (1.60% ± 0.18%) (Figure 1E). However, the percentages of thiazole orange–positive platelets in Agk−/− mice (1.07% ± 0.53%) were obviously lower than those in Agkf/f mice (2.45% ± 1.03%) (Figure 1F).
AGK deficiency caused inefficient BM thrombopoiesis and excessive extramedullary hematopoiesis
To understand the cause of thrombocytopenia in the Agk−/− mice, megakaryocytes in BM and spleen were stained with anti-CD42c antibodies. There were 20% fewer megakaryocytes in Agk−/− mouse femur sections (4.168 ± 0.1265/mm2) than in Agkf/f mouse femur sections (5.136 ± 0.1388/mm2). However, there were fourfold more megakaryocytes in Agk−/− mouse spleen sections (1.619 ± 0.1537/mm2) than in Agkf/f mouse spleen sections (0.4286 ± 0.05719/mm2) (Figure 2A). Next, the percentages of CD41+CD42b+ megakaryocytes in BM cells and spleen cells of Agk−/− mice and Agkf/f mice were measured by flow cytometry. The BM of AGK-deficient mice had 6.102 ± 0.2419% CD41+CD42b+ cells, while that of Agkf/f mice had 10.01 ± 0.7500% CD41+CD42b+ cells; therefore, Agk−/− BM exhibited nearly 40% fewer megakaryocytes than Agkf/f BM. The spleens of Agk−/− mice had 9.094 ± 0.6412% megakaryocytes, whereas those of Agkf/f mice had 6.078 ± 0.3065% megakaryocytes (Figure 2B). In addition, Agk−/− mice developed splenomegaly compared with Agkf/f mice (Figure 2C). However, there was no obvious morphological alteration of BM and splenic megakaryocytes in Agk−/− mice (supplemental Figure 1D). Meanwhile, the proportion of major cells in BM and spleen was not changed in both Agkf/f and Agk−/− mice, except for a slight increase in neutrophils in the spleens of Agk−/− mice (supplemental Figure 1E-F). It has been reported that the increase in splenic megakaryocytes may be explained by compensatory hematopoiesis.34,35 To determine whether AGK deficiency causes compensatory splenomegaly, the spleens of Agkf/f and Agk−/− mice were removed and platelet counts were monitored. In Agkf/f mice, platelet count increased up to day 10 after splenectomy and gradually decreased thereafter, returning to preoperative levels by day 50. Platelet counts in Agk−/− mice progressively decreased from day 20 to day 50 postsplenectomy, and by day 50, counts were much lower than preoperative values (Figure 2D). A BM transplantation assay showed that the splenomegaly and thrombocytopenia in Agk−/− mice were transplantable (supplemental Figure 2C-D). These results demonstrated that AGK deficiency causes inefficient thrombopoiesis and excessive extramedullary hematopoiesis.
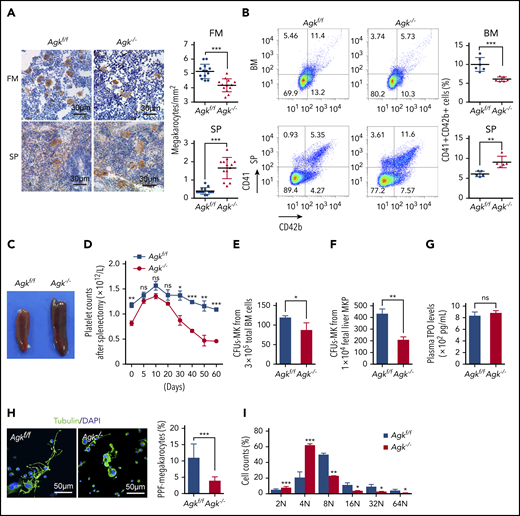
Impaired megakaryocyte development in Agkf/fPF4-Cre mice. (A) Immunohistochemical images of CD42c in femurs and spleens of Agkf/f mice and Agk−/− mice. The scale bars represent 30 μm. Statistics for the megakaryocytes in femur (FM) and spleen (SP) sections of Agkf/f mice and Agk−/− mice are shown (n = 13; ***P < .001). (B) CD41+CD42b+ megakaryocytes in BM and spleen samples were detected by flow cytometry. Statistics for the CD41+CD42b+ megakaryocytes in the BM (n = 6; ***P < .001) and spleen (n = 5; **P < .01) samples of Agkf/f mice and Agk−/− mice are shown. (C) Spleens of Agkf/f mice and Agk−/− mice. (D) Splenectomies were performed on Agkf/f mice and Agk−/− mice, and circulating platelet were counted at indicated time points (n = 5; *P < .05, **P < .01, ***P < .001). (E) Megakaryocyte colony-forming unit (MK-CFU) assay using BM cells harvested from Agkf/f mice and Agk−/− mice (n = 3; *P < .05). (F) Megakaryocyte colony-forming unit assay using sorted MKP cells from fetal liver of Agkf/f mice and Agk−/− mice (n = 3; **P < .01). (G) The level of TPO in the plasma from Agkf/f mice and Agk−/− mice (n = 6). (H) Immunofluorescence images of α-tubulin in PPF-megakaryocytes derived from the fetal livers of Agkf/f mice and Agk−/− mice. Scale bars, 50 μm. Statistics for the PPF-megakaryocytes/total megakaryocytes derived from the fetal livers of Agkf/f mice and Agk−/− mice are shown (n = 10; ***P < .001). (I) Percentage of megakaryocytes polyploidy derived from fetal liver of Agkf/f and Agk−/− mice (n = 3; *P < .05, **P < .01, ***P < .001). DAPI, 4′,6-diamidino-2-phenylindole; MK, megakaryocyte; MKP, megakaryocyte progenitor.
Impaired megakaryocyte development in Agkf/fPF4-Cre mice. (A) Immunohistochemical images of CD42c in femurs and spleens of Agkf/f mice and Agk−/− mice. The scale bars represent 30 μm. Statistics for the megakaryocytes in femur (FM) and spleen (SP) sections of Agkf/f mice and Agk−/− mice are shown (n = 13; ***P < .001). (B) CD41+CD42b+ megakaryocytes in BM and spleen samples were detected by flow cytometry. Statistics for the CD41+CD42b+ megakaryocytes in the BM (n = 6; ***P < .001) and spleen (n = 5; **P < .01) samples of Agkf/f mice and Agk−/− mice are shown. (C) Spleens of Agkf/f mice and Agk−/− mice. (D) Splenectomies were performed on Agkf/f mice and Agk−/− mice, and circulating platelet were counted at indicated time points (n = 5; *P < .05, **P < .01, ***P < .001). (E) Megakaryocyte colony-forming unit (MK-CFU) assay using BM cells harvested from Agkf/f mice and Agk−/− mice (n = 3; *P < .05). (F) Megakaryocyte colony-forming unit assay using sorted MKP cells from fetal liver of Agkf/f mice and Agk−/− mice (n = 3; **P < .01). (G) The level of TPO in the plasma from Agkf/f mice and Agk−/− mice (n = 6). (H) Immunofluorescence images of α-tubulin in PPF-megakaryocytes derived from the fetal livers of Agkf/f mice and Agk−/− mice. Scale bars, 50 μm. Statistics for the PPF-megakaryocytes/total megakaryocytes derived from the fetal livers of Agkf/f mice and Agk−/− mice are shown (n = 10; ***P < .001). (I) Percentage of megakaryocytes polyploidy derived from fetal liver of Agkf/f and Agk−/− mice (n = 3; *P < .05, **P < .01, ***P < .001). DAPI, 4′,6-diamidino-2-phenylindole; MK, megakaryocyte; MKP, megakaryocyte progenitor.
AGK deficiency significantly impaired megakaryocyte development and differentiation
Megakaryocytes are derived from HSCs, which undergo complex processes, including commitment of HSCs and the proliferation and terminal differentiation of MKPs.36 To explore whether AGK is involved in the proliferation and early differentiation of MKPs, a CFU-MK assay was performed. Results in Figure 2E-F showed that both total BM cells and fetal liver–derived MKPs from Agk−/− mice formed fewer CFU-MKs (86.67 ± 18.77 in BM; 208.7 ± 13.91 in MKPs) than those from Agkf/f mice (119.0 ± 4.58 in BM; 430.7 ± 23.39 in MKPs), demonstrating that Agk−/− MKPs had decreased response to exogenous TPO and exhibited block of terminal differentiation. There was no difference of plasma TPO levels between Agkf/f and Agk−/− mice (Figure 2G), further indicating that AGK could regulate terminal differentiation of megakaryocyte instead of TPO production. Also, the protein levels of AGK in Agk−/− megakaryocytes and MKPs were reduced compared with those in Agkf/f mice (supplemental Figure 1G-H).
To further explore the roles of AGK in megakaryocyte differentiation, fetal livers were isolated from Agkf/f and Agk−/− embryos and homogenized into single cells. There was no difference in the proportion of CD41+ cell population in the steady state of fetal liver of Agkf/f and Agk−/− mice (supplemental Figure 3B). After fetal liver cells were incubated with IL-3 and TPO for 5 days, BSA gradient separation was performed to obtain megakaryocytes. PPF experiments showed that AGK-deficient megakaryocytes generated fewer proplatelets than control megakaryocytes (Figure 2H). Also, Agk−/− megakaryocytes were morphologically less differentiated than Agkf/f megakaryocytes (Figure 2H). Ploidy examination revealed that compared with Agkf/f megakaryocytes, Agk−/− megakaryocytes included more cells arrested in the tetraploid phase (4N) but fewer cells in higher ploidy phases (≥8N) (Figure 2I). These results suggest that AGK deficiency impairs megakaryocyte development.
Megakaryocytes could be a niche for HSC development.37,38 The hematopoietic populations upstream of megakaryocytes, including long-term HSCs, short-term HSCs, multipotent progenitors, CD41+ HSCs, common myeloid progenitors, megakaryocyte and erythroid progenitors, granulocyte and macrophage progenitors, and MKPs in the BM, were assessed by flow cytometry. There was no alternation of the populations upstream of the megakaryocytes in the BM of Agk−/− mice compared with that of Agkf/f mice (supplemental Figure 2A-B), suggesting that AGK deficiency in megakaryocytes probably only affected the differentiation of HSCs into megakaryocytes.
AGK regulated thrombocytopoiesis through a nonmitochondrial function
AGK is a kinase and plays an important role in mitochondrial homeostasis maintenance. It has been reported that the G126E mutation suppresses AGK kinase activity by abolishing adenosine triphosphate binding. To elucidate the roles of AGK kinase activity in thrombocytopoiesis, AGK G126E point-mutant (PM) mice were generated as described in our previous research (Figure 3A).39 The levels of AGK did not differ between PM mice and wild-type (WT) mice (Figure 3B). The blood count results demonstrated that WT and PM mice had similar counts of platelets and other blood cells (Figure 3C). WT and PM megakaryocytes were prepared, and PPF experiments were performed. The results in Figure 3D demonstrate that the AGK G126E mutation had no effect on the numbers or morphology of megakaryocyte-derived proplatelets. The OCRs of the platelets of PM and Agk−/− mice were measured to further elucidate the mitochondrial roles of AGK in megakaryocyte differentiation. The results presented in Figure 3E-F show that there were no differences in platelet oxygen consumption between WT and PM mice or between Agk−/− and Agkf/f mice. Thus, we propose that AGK regulates thrombocytopoiesis through a nonmitochondrial function.
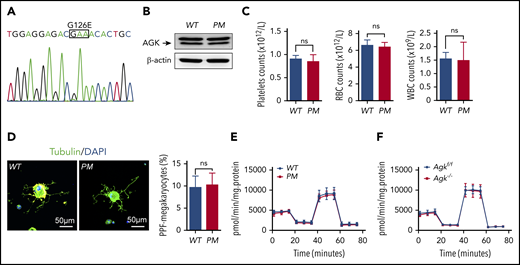
AGK regulates thrombocytopoiesis through a nonmitochondrial function. (A) Genotyping of AGK-G126E PM mice by sequencing. (B) The levels of AGK in the platelets of AGK-G126E PM mice were tested by western blot analysis. (C) Platelet, RBC, and WBC counts in WT and AGK-G126E PM mice (n = 4). (D) Immunofluorescence images of α-tubulin in PPF-megakaryocytes derived from the fetal livers of WT mice and AGK-G126E PM mice. The scale bars represent 50 μm. Statistics for the PPF-megakaryocytes/total megakaryocytes derived from the fetal livers of WT mice and AGK-G126E PM mice are shown (n = 10). (E) Extracellular flux analysis of the OCRs of the platelets of WT and PM mice; 2 μM oligomycin, 0.25 μM FCCP, and 1 μM rotenone/antimycin A were added at the indicated time points. The OCR was normalized to the protein amount (n = 6). (F) Extracellular flux analysis of the OCRs of the platelets of Agkf/f and Agk−/− mice; 2 μM oligomycin, 0.25 μM FCCP, and 1 μM rotenone/antimycin A were added at the indicated time points. The OCR was normalized to the protein amount (n = 4).
AGK regulates thrombocytopoiesis through a nonmitochondrial function. (A) Genotyping of AGK-G126E PM mice by sequencing. (B) The levels of AGK in the platelets of AGK-G126E PM mice were tested by western blot analysis. (C) Platelet, RBC, and WBC counts in WT and AGK-G126E PM mice (n = 4). (D) Immunofluorescence images of α-tubulin in PPF-megakaryocytes derived from the fetal livers of WT mice and AGK-G126E PM mice. The scale bars represent 50 μm. Statistics for the PPF-megakaryocytes/total megakaryocytes derived from the fetal livers of WT mice and AGK-G126E PM mice are shown (n = 10). (E) Extracellular flux analysis of the OCRs of the platelets of WT and PM mice; 2 μM oligomycin, 0.25 μM FCCP, and 1 μM rotenone/antimycin A were added at the indicated time points. The OCR was normalized to the protein amount (n = 6). (F) Extracellular flux analysis of the OCRs of the platelets of Agkf/f and Agk−/− mice; 2 μM oligomycin, 0.25 μM FCCP, and 1 μM rotenone/antimycin A were added at the indicated time points. The OCR was normalized to the protein amount (n = 4).
AGK maintained JAK2/Stat3 pathway activation during megakaryocyte differentiation
To further elucidate the mechanism, the location of AGK was first analyzed in HEK293T and Meg01 cells transfected with plasmids expressing AGK-Flag. We found that AGK localized to the cytoplasm in addition to mitochondria (Figure 4A; supplemental Figure 3C).

AGK facilitates the JAK2/Stat3 signaling pathway in platelets and megakaryocytes by binding to JAK2. (A) Immunofluorescence image of HEK293T cells transfected with AGK-Flag. HEK293T cells were stained with Flag antibodies (Alexa Fluor 488), DAPI, and a mitochondria-targeting dye (MitoTracker Deep Red). The scale bars represent 30 μm. (B) Coimmunoprecipitation of the lysates of HEK293T cells transfected with JAK2-HA and AGK-Flag/AGK-G126E-Flag using HA beads or Flag beads. (C) Immunoprecipitation of platelet lysates with anti-AGK or anti- JAK2 antibodies. (D) The JAK2 and Stat3 phosphorylation levels of WT and AGK-null platelets in response to 25 IU/mL TPO were tested by western blot analysis. (E) The JAK2 and Stat3 phosphorylation levels of the platelets of WT and AGK-G126E PM mice in response to 25 IU/mL TPO were tested by western blot analysis. (F) Immunofluorescence images of undifferentiated or differentiated megakaryocytes derived from the fetal livers of Agkf/f and Agk−/− mice. Megakaryocytes were stained with tubulin antibodies (Alexa Fluor 488), DAPI, and Stat3 antibodies (rhodamine). Scale bars, 50 μm. IgG, immunoglobulin G; IP, immunoprecipitation.
AGK facilitates the JAK2/Stat3 signaling pathway in platelets and megakaryocytes by binding to JAK2. (A) Immunofluorescence image of HEK293T cells transfected with AGK-Flag. HEK293T cells were stained with Flag antibodies (Alexa Fluor 488), DAPI, and a mitochondria-targeting dye (MitoTracker Deep Red). The scale bars represent 30 μm. (B) Coimmunoprecipitation of the lysates of HEK293T cells transfected with JAK2-HA and AGK-Flag/AGK-G126E-Flag using HA beads or Flag beads. (C) Immunoprecipitation of platelet lysates with anti-AGK or anti- JAK2 antibodies. (D) The JAK2 and Stat3 phosphorylation levels of WT and AGK-null platelets in response to 25 IU/mL TPO were tested by western blot analysis. (E) The JAK2 and Stat3 phosphorylation levels of the platelets of WT and AGK-G126E PM mice in response to 25 IU/mL TPO were tested by western blot analysis. (F) Immunofluorescence images of undifferentiated or differentiated megakaryocytes derived from the fetal livers of Agkf/f and Agk−/− mice. Megakaryocytes were stained with tubulin antibodies (Alexa Fluor 488), DAPI, and Stat3 antibodies (rhodamine). Scale bars, 50 μm. IgG, immunoglobulin G; IP, immunoprecipitation.
It has been reported that AGK can bind to the JAK2 JH2 domain and amplify JAK2/Stat3 signaling in esophageal squamous cells.40 The JAK2/Stat3 signaling pathway is vital in the differentiation and development of myeloid cells. Thus, it was necessary to verify whether AGK regulated megakaryocyte development through the JAK2/Stat3 signaling pathway.
Immunoprecipitation was performed, and the results showed that AGK could associate with JAK2 in both HEK293T cells and platelets (Figure 4B-C). Compared with control platelets, the phosphorylation levels of JAK2 and Stat3 were suppressed in AGK-null platelets treated with 25 IU/mL TPO (Figure 4D). Meanwhile, the phosphorylation levels of JAK2 and Stat3 were also suppressed in MKs of both BM and spleen from Agk−/− mice compared with Agkf/f mice in response to 25 IU/mL TPO (supplemental Figure 1I-J). WT and PM platelets were then stimulated with TPO. There were no differences in the phosphorylation levels of JAK2 and Stat3 between WT and PM platelets in response to 25 IU/mL TPO (Figure 4E). Immunoprecipitation of HEK293T cells transfected with plasmids expressing JAK2-HA, AGK-Flag, or G126E-Flag also illustrated that the AGK G126E mutation had no effect on JAK2/AGK binding (Figure 4B). These results demonstrate that JAK2/Stat3 signaling is independent of AGK kinase activity.
The immunofluorescence staining results showed that Stat3 was distributed in the nucleus and cytoplasm of Agkf/f megakaryocytes (Figure 4F; supplemental Figure 3A) and has no colocalization with α-granule (supplemental Figure 3D). However, AGK deficiency suppressed Stat3 entry into the nucleus in both undifferentiated and differentiated megakaryocytes. These results imply that AGK maintains JAK2/Stat3 signaling and that deletion of AGK impairs megakaryocyte differentiation.
The peptide YGVCFCGDENI (617F) promoted AGK binding to JAK2
The kinase activity of JAK2 is regulated by the interaction of 2 C-terminal JAK homology domains (JH1 and JH2). The JH1 domain is responsible for the phosphorylation of the target tyrosine (Y) residue of the receptor, which activates downstream signaling molecules containing SH2 domains, including Stat family and other signaling molecules. The JH2 domain is regarded as a pseudokinase domain that abrogates the function of the JH1 domain under physiological conditions.41 The V617F mutation in the JAK2 JH2 domain has been found in most cases of MPNs, including PV, ET, and PMF.42
A previous study indicated that AGK binds to the JAK2 JH2 domain to remove the inhibitory effect of this domain on the catalytically active JH1 domain and thus promote the activation of the JAK2/Stat3 signaling pathway.40 To explore the function of AGK in JAK2 V617F mutation–induced hyperactivation of JAK2/Stat3 signaling, HEK293T cells were first transfected with AGK-Flag, JAK2-HA, or JAK2 V617F-HA plasmids. The immunoprecipitation results demonstrated that JAK2 V617F promoted the interaction between AGK and JAK2 (Figure 5A).
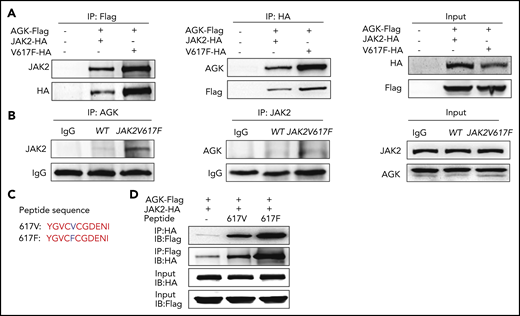
Peptide 617F (YGVCFCGDENI) facilitates the binding of AGK and JAK2. (A) Coimmunoprecipitation of the lysates of HEK293T cells transfected with AGK-Flag and JAK2-HA/JAK2-V617F-HA using HA beads or Flag beads. (B) Immunoprecipitation of the lysates of platelet from WT mice and JAK2V617F mice with anti-AGK or anti-JAK2 antibodies. (C) Sequences of peptides 617V and 617F. (D) Coimmunoprecipitation of the lysates of HEK293T cells transfected with AGK-Flag and JAK2-HA and incubated with or without peptide 617F/617V using HA beads or Flag beads. IB, immunoblot.
Peptide 617F (YGVCFCGDENI) facilitates the binding of AGK and JAK2. (A) Coimmunoprecipitation of the lysates of HEK293T cells transfected with AGK-Flag and JAK2-HA/JAK2-V617F-HA using HA beads or Flag beads. (B) Immunoprecipitation of the lysates of platelet from WT mice and JAK2V617F mice with anti-AGK or anti-JAK2 antibodies. (C) Sequences of peptides 617V and 617F. (D) Coimmunoprecipitation of the lysates of HEK293T cells transfected with AGK-Flag and JAK2-HA and incubated with or without peptide 617F/617V using HA beads or Flag beads. IB, immunoblot.
To explore the binding of AGK and JAK2 in endogenous JAK2 V617F mutation, a transgenic mouse model with vav promoter–driven JAK2V617F expression was used.43 Platelets from JAK2V617F transgenic mice and WT mice were prepared, and immunoprecipitation assays were carried out. The results in Figure 5B confirmed that endogenous JAK2 V617F also promoted AGK binding to JAK2.
Crystal structure analysis showed that only Phe594, Phe595, and Phe617 exhibited rotations or shifts in their spatial positions in the V617F-mutant JH2 domain compared with the WT JH2 domain.44 This led us to wonder how such small changes could so greatly increase the kinase activity. The peptides YGVCVCGDENI (617V) and YGVCFCGDENI (617F) were synthesized (Figure 5C), and immunoprecipitation experiments were carried out to resolve this issue. The results in Figure 5D show that both the peptides (671F and 617V) increased the AGK and JAK2 interaction, but peptide 617F enhanced AGK binding to JAK2 to a greater extent than did peptide 617V in HEK293T cell lysates.
The peptide YGVCFCGDENI (617F) accelerated megakaryocyte differentiation into proplatelets
The use of cell-penetrating peptides (CPPs) is an efficient method to help macromolecules penetrate the cell membrane. The peptide TAT(GRKKRRQRRR) from HIV has been reported to be able to transport large molecular carriers into cells.45 To study the biological ramifications of the promoting effects of the peptides 617V and 617F, we further synthesized versions of these 2 peptides (C-617V and C-617F) with CPPs (TAT) on the N-terminal (Figure 6A). Immunofluorescence images of HEK293T cells treated with the TAMRA-labeled C-617F or C-671V peptide showed that these 2 peptides could successfully penetrate into cells (Figure 6B). The effects of peptides C-617F and C-617V on JAK2/Stat3 signaling in Agkf/f and Agk−/− platelets were further studied. We found that both peptides augmented the JAK2/Stat3 signaling pathway in platelets, but peptide C-617F facilitated JAK2/Stat3 signaling more strongly than did peptide C-617V (Figure 6C). In addition, the promoting effects of peptide C-617F were mostly dependent on the presence of AGK (Figure 6D). These results suggested that peptide C-617F was able to facilitate JAK2/Stat3 signaling greatly in platelets.
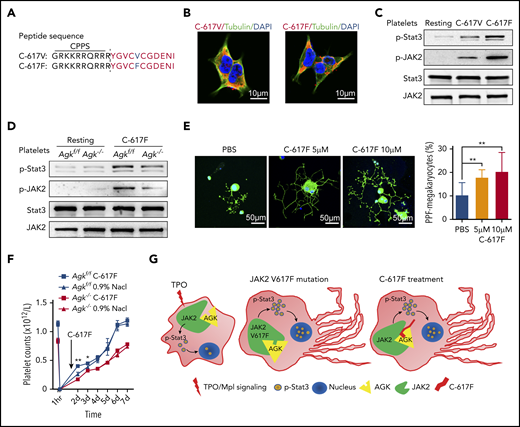
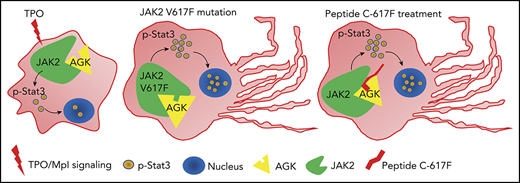
Peptide C-617F accelerates megakaryocyte PPF. (A) Sequences of C-617V and C-617F peptides with N-terminal CPPs. (B) Immunofluorescence images of α-tubulin in HEK293T cells treated with TAMRA-labeled C-617F or TAMRA-labeled C-617V. Scale bars, 10 μm. (C) Immunoblot analysis of Stat3 and JAK2 phosphorylation levels in platelets incubated with or without 10 μM peptide C-617F/C-617V for 20 minutes at 37°C. (D) Immunoblot analysis of Stat3 phosphorylation levels in WT or AGK-null platelets incubated with or without 10 μM peptide C-617F for 20 min at 37°C. (E) Immunofluorescence images of fetal liver–derived megakaryocytes stimulated with PBS, 5 μM peptide C-617F, or 10 μM peptide C-617F and stained with α-tubulin antibodies (Alexa Fluor 488) and DAPI. Scale bars, 50 μm. Statistics for the PPF-megakaryocytes/total megakaryocytes among megakaryocytes stimulated with PBS, 5 μM peptide C-617F, or 10 μM peptide C-617F are shown (n = 10, **P < .01). (F) Platelets of Agkf/f and Agk−/− mice were eliminated by tail IV injection of anti-CD42b (2 µg/g). BM injection of the peptide C-617F (20 µg/g) was carried out 36 hours later, and platelet counts were monitored with a HEMAVET automated hematologic analyzer at different time points (n = 3; *P < .05, **P < .01). (G) Schematic of peptide 617F-mediated augmentation of JAK2/Stat3 signaling resulting from increased interaction between AGK and JAK2 in megakaryocytes.
Peptide C-617F accelerates megakaryocyte PPF. (A) Sequences of C-617V and C-617F peptides with N-terminal CPPs. (B) Immunofluorescence images of α-tubulin in HEK293T cells treated with TAMRA-labeled C-617F or TAMRA-labeled C-617V. Scale bars, 10 μm. (C) Immunoblot analysis of Stat3 and JAK2 phosphorylation levels in platelets incubated with or without 10 μM peptide C-617F/C-617V for 20 minutes at 37°C. (D) Immunoblot analysis of Stat3 phosphorylation levels in WT or AGK-null platelets incubated with or without 10 μM peptide C-617F for 20 min at 37°C. (E) Immunofluorescence images of fetal liver–derived megakaryocytes stimulated with PBS, 5 μM peptide C-617F, or 10 μM peptide C-617F and stained with α-tubulin antibodies (Alexa Fluor 488) and DAPI. Scale bars, 50 μm. Statistics for the PPF-megakaryocytes/total megakaryocytes among megakaryocytes stimulated with PBS, 5 μM peptide C-617F, or 10 μM peptide C-617F are shown (n = 10, **P < .01). (F) Platelets of Agkf/f and Agk−/− mice were eliminated by tail IV injection of anti-CD42b (2 µg/g). BM injection of the peptide C-617F (20 µg/g) was carried out 36 hours later, and platelet counts were monitored with a HEMAVET automated hematologic analyzer at different time points (n = 3; *P < .05, **P < .01). (G) Schematic of peptide 617F-mediated augmentation of JAK2/Stat3 signaling resulting from increased interaction between AGK and JAK2 in megakaryocytes.
The effects of peptide C-617F on thrombocytopoiesis were further examined. Specifically, fetal liver–derived megakaryocytes were incubated with different doses of peptide C-617F. Compared with phosphate-buffered saline (PBS) treatment, peptide C-617F treatment resulted in longer and more branched megakaryocyte morphologies (Figure 6E). Statistical analysis showed that peptide C-617F significantly facilitated the differentiation of megakaryocytes into proplatelets (Figure 6E).
To study the effects of peptide C-617F on megakaryopoiesis in vivo, peptides were directly injected in BM and platelet counts were measured at different time points. The results in Figure 6F showed that peptide C-617F could significantly increase the platelet counts in Agkf/f mice at 12h after the injection of peptide, but the peptide had no enhancing effects on platelet counts in Agk−/− mice. Therefore, peptide C-617F could enhance platelet counts in vivo at an AGK-dependent manner (Figure 6G).
Discussion
AGK was initially identified as a mitochondrial inner membrane protein that phosphorylates diacylglycerol or monoacylglycerol to produce phosphatidic acid or lysophosphatidic acid, respectively.20,21 Recently, several studies have also suggested that AGK is a subunit of the TIM22 complex that helps import mitochondrial carrier proteins, suggesting that AGK has kinase-independent and kinase-dependent mitochondrial functions.22-24 Our study demonstrated that ablation of AGK kinase activity (through G126E mutation) did not affect peripheral platelet counts or megakaryocyte differentiation, suggesting that the mitochondrial kinase function of AGK is not required for thrombocytopoiesis. The OCRs of Agk−/− platelets and AGK G126E mutant platelets did not differ from those of control platelets, further implying that AGK is probably not important for megakaryocyte/platelet mitochondrial function. Our study recently reported that cytosolic AGK interacts with phosphatase and tensin homolog (PTEN) and triggers PTEN phosphorylation, thereby restricting PTEN phosphatase activity in CD8+ T cells.39 All of these results suggest that AGK exhibits diverse functions in specific cells via distinct mechanisms.
The Mpl/JAK2/Stat3 pathway is the major signaling pathway involved in the regulation of megakaryocyte development.46,47 Our study confirmed that cytosolic AGK bound to JAK2 in megakaryocytes/platelets. The association of AGK with JAK2 significantly enhanced TPO/Mpl-mediated JAK2/Stat3 activation in megakaryocytes, thereby promoting thrombocytopoiesis. The JAK2/Stat3 pathway has been reported to positively regulate agonist-induced platelet aggregation, calcium mobilization, and platelet plug formation, indicating that AGK probably plays a role in agonist-induced platelet activation and thrombus formation, which needs further studies.48
ET, together with PV and PMF, is described as classic Philadelphia chromosome–negative MPN. MPNs are clonal disorders of hematopoietic progenitors that result in disruption of the hematopoietic system and increased risks of thrombosis and leukemia.13,14,42 Mutant alleles associated with MPNs have been discovered in the past 3 decades; the majority of cases are driven by JAK2, Mpl, and CALR mutations that result in continuous activation of JAK/Stat signaling.12,13 JAK2 V617F is a frequent mutation in patients with MPNs. Therefore, several JAK2 inhibitors have been successfully applied for the treatment of MPNs or other diseases.49,50 The JAK2 JH2 domain is an autoinhibitory domain that controls the kinase activity of JAK2 under physiological conditions. It is believed that the JAK2 V617F mutation in the JH2 domain abolishes the inhibitory effect of this domain, leading to the continuous activation of JAK2/Stat3 signaling and hyperactivation of the hematopoietic system.13,41 In our study, we found that the JAK2 V617F mutation dramatically enhanced JAK2 binding to AGK and greatly facilitated JAK2/Stat3 signaling in megakaryocytes/platelets in response to TPO in vitro and in vivo. In addition, AGK deficiency greatly suppressed TPO-induced activation of JAK2/Stat3 signaling. All of these results suggest that targeting the binding sites of JAK2 and AGK to inhibit their association may be a new strategy for inhibition of JAK2 activity and treatment of MPNs. However, the cocrystal structure must be elucidated before an inhibitor blocking JAK2 and AGK binding can be developed.
Thrombocytopenia is a kind of severe disease associated with increased risk of vascular, mucosal and visceral hemorrhage.51 Thrombocytopenia is caused by impaired production of platelets from megakaryocytes (as occurs in aplastic anemia and myelofibrosis) or hyperactive destruction of platelets (as occurs in immune thrombocytopenia purpura and thrombotic thrombocytopenia purpura).52,53 Most drugs for the treatment of thrombocytopenia are TPO or Mpl agonists.15,16 In our study, we found that the peptide YGVCF617CGDENI mimicking the JAK2 V617F mutation enhanced the binding of AGK to JAK2 and that the cell-permeable peptide C-617F containing the YGVCF617CGDENI sequence significantly augmented JAK2/Stat3 signaling, accelerated PPF, and enhanced platelet counts in vivo. These results indicate that the peptide C-617F or small-molecule analogs enhancing AGK binding to JAK2 may be novel therapeutic agents for thrombocytopenia.
Therefore, our study reveals critical roles of AGK in megakaryocyte differentiation and platelet biogenesis and suggests that targeting the interaction between AGK and JAK2 may be a novel strategy for the treatment of thrombocytopenia and thrombocythemia.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Kevin Liu from Shanghai High School International Division for proofreading the article.
This work was supported by the National Natural Science Foundation of China (grants 81525001, 91739302, 31830050, and 81721004 to J.L.; grant 81900138 to Y.X.; grant 81800129 to Lin Zhang; grants 81600104 and 81970123 to X.F.; and grants 81970121 and 81770128 to Lei Zhang) and the China Postdoctoral Science Foundation (grant 2019M651528 to Y.X.).
Authorship
Contribution: H.J., Y.X., and J.L. designed the experiments, analyzed data, and wrote the paper; H.J., Y.X., and Z.Y. performed the experiments; and N.D., M.Y., Lin Zhang, X.F., Y.Z., Q.Z., J.H., Lei Zhang, and J.Z. helped with the experiments.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Junling Liu, Department of Biochemistry and Molecular Cell Biology, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Rd, Shanghai 200025, China; e-mail: liujl@shsmu.edu.cn; Yanyan Xu, Department of Biochemistry and Molecular Cell Biology, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao Tong University School of Medicine, 280 South Chongqing Rd, Shanghai 200025, China; e-mail: xuyanyan901@shsmu.edu.cn; and Lei Zhang, State Key Laboratory of Experimental Hematology, Institute of Hematology & Blood Diseases Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, 288 Nanjing Rd, Tianjin 3000200, China; e-mail: zhanglei1@ihcams.ac.cn.