Key Points
Insertion of ASS or OTC enzymes enhances CAR-T cell proliferation and activity in the low arginine microenvironment.

Abstract
Hematological and solid cancers catabolize the semiessential amino acid arginine to drive cell proliferation. However, the resulting low arginine microenvironment also impairs chimeric antigen receptor T cells (CAR-T) cell proliferation, limiting their efficacy in clinical trials against hematological and solid malignancies. T cells are susceptible to the low arginine microenvironment because of the low expression of the arginine resynthesis enzymes argininosuccinate synthase (ASS) and ornithine transcarbamylase (OTC). We demonstrate that T cells can be reengineered to express functional ASS or OTC enzymes, in concert with different chimeric antigen receptors. Enzyme modifications increase CAR-T cell proliferation, with no loss of CAR cytotoxicity or increased exhaustion. In vivo, enzyme-modified CAR-T cells lead to enhanced clearance of leukemia or solid tumor burden, providing the first metabolic modification to enhance CAR-T cell therapies.
Introduction
Chimeric antigen receptor T cells (CAR-T) cells are patient-derived T cells engineered with an antibody fragment (scFv) against target cell surface antigens. Proof-of-principle was established in patients with relapsed pediatric B-cell acute lymphoblastic leukemia who underwent sustained remissions using anti-CD19 CAR-T cells.1 However, in hematological cancers such as acute myeloid leukemia (AML) or solid tumors such as neuroblastoma, mesothelioma, or glioblastomas, only limited, temporary antitumor efficacy has been demonstrated.2,3 In the majority of clinical trials, CAR-T cells became undetectable within days to weeks. These findings suggest that the tumor microenvironment impairs expansion of CAR-T cells. In these trials, persistence of CAR-T cells correlated with a longer survival; thus, strategies to enhance cellular proliferation could improve outcome. Here we studied reengineering cell metabolism to improve CAR-T cell activity.
Methods
Vectors and CAR-T constructs
To generate T cells expressing the CAR for GD2, a second-generation CAR plasmid containing the already-established anti-GD2-ScFv-41BB-CD3ζ expression cassette downstream of a truncated CD34 tag and separated by a self-cleaving F2A peptide, was synthesized by GeneScript.4 The anti-GD2-ScFv (derived from the 14.G2a chimeric immunoglobulin G2a anti-GD2 monoclonal antibody) and 41BB domains of the expression cassette are separated by a CH2CH3 spacer region as well as the hinge and transmembrane domains of CD8α. To generate anti-CD33 (Siglec-3), anti-mesothelin, and anti-EGFRvIII-specific CARs, respective scFv coding sequences were added to the 41BB-CD3ζ expression cassette. For enzyme modification of the base CAR sequences, Insert sequences comprising the full codon optimized coding sequences of human argininosuccinate synthase (ASS) and/or human ornithine transcarbamylase (OTC) enzymes were synthesized by GeneScript and subcloned downstream of and within the same open reading frame of the base CAR expression cassettes, with each coding sequence separated by a self-cleaving P2A peptide. All sequences were validated by DNA sequencing.
Enzyme-linked immunosorbent assay
The concentrations of arginine in plasma from patients and tumor bearing mice were quantified using a competitive enzyme linked immunoassay (Immunodiagnostik K7733) according to the manufacturer’s instructions. Concentrations of interferon-γ released from T cells were measured by enzyme-linked immunosorbent assay according to the manufacturer’s instructions (BioLegend).
Results and discussion
Previously, we showed that a low arginine microenvironment suppresses antigen-specific or CAR-T cell responses, by providing a brake on T-cell proliferation.4-7 Peripheral blood arginine concentrations are lower than healthy controls in AML, neuroblastoma, and pediatric cancer patients at diagnosis (Figure 1A-C) and Arginase I is expressed within the tumor microenvironments of adult tumor subtypes (supplemental Figure 1A-B, available on the Blood Web site).8 T cells are acutely sensitive to extracellular concentrations of arginine because they have low/absent expression of the arginine resynthesis pathway enzymes ASS and OTC (supplemental Figures 1C and 2A).9-12


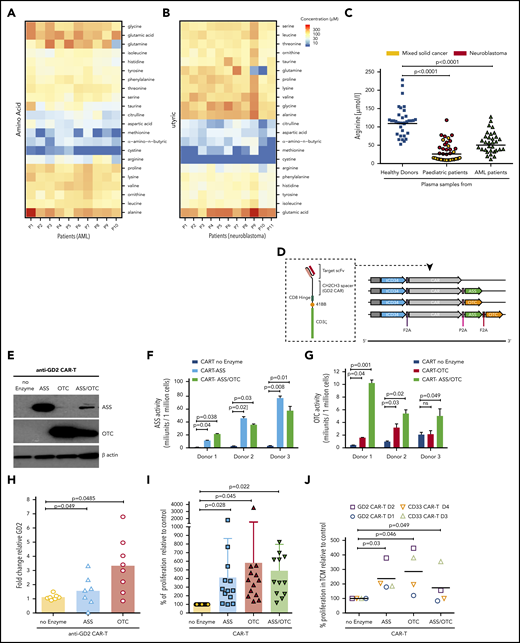
Insertion of ASS and OTC enzymes enhances CAR-T cell proliferation in vitro. Biochrom amino acid analysis of (A) AML (n = 10) and (B) neuroblastoma (n = 11) plasma samples revealing low concentrations of arginine, cysteine, methionine, a-amino-n-butyric, aspartic acid, citrulline, and taurine at diagnosis. (C) Plasma arginine levels at diagnosis are significantly lower than healthy controls in AML and solid pediatric cancer patients. (D) Schematic of CAR-T constructs containing the basic anti-XX-CAR scFv-CD8 hinge-41BB-CD3ζ in concert with ASS or OTC or ASS+OTC enzyme. A truncated CD34 is expressed for CAR-T identification and purification. (E) Western blot of ASS and OTC expression, with β-actin control, in control and modified anti-GD2-CAR-T cells posttransduction and expansion. (Representative of n = 7.) (F) ASS enzyme activity, measured by citrulline catabolism, is increased in ASS expressing CAR-T cells compared with controls. (G) OTC enzyme activity, measured by citrulline production, is increased in OTC expressing CAR-T cells compared with controls. (H) Highest proliferation of 2.5 × 106 modified anti-GD2-Jurkat CAR-T cell line following engraftment into NOG-SCID mice treated with recombinant human arginase (BCT-100, daily 50 mg/kg IV). (I) Proliferation of modified human CAR-T cells is significantly increased in low arginine media conditions, in vitro, as measured by flow cytometry after 72 hours. (J) Proliferation of modified anti-GD2 or anti-CD33-CAR-T cells is significantly increased in neuroblastoma (LAN-1 neuroblastoma or K562 AML, 72-hour conditioned supernatants) conditions, in vitro as measured by flow cytometry after 72 hours. (K) Expansion of modified CAR-T cells is enhanced in low arginine vs excess arginine (RPMI10%) conditions as measured by flow cytometry after 72 hours. (L) Liquid chromatography mass spectometry analysis of modified CAR-T cells after proliferation in low arginine conditions (72 hours) compared with each modified version. Heatmap demonstrating metabolomic changes in arginine and proline metabolism, pyrimidine metabolism, and purine metabolism pathways.
Insertion of ASS and OTC enzymes enhances CAR-T cell proliferation in vitro. Biochrom amino acid analysis of (A) AML (n = 10) and (B) neuroblastoma (n = 11) plasma samples revealing low concentrations of arginine, cysteine, methionine, a-amino-n-butyric, aspartic acid, citrulline, and taurine at diagnosis. (C) Plasma arginine levels at diagnosis are significantly lower than healthy controls in AML and solid pediatric cancer patients. (D) Schematic of CAR-T constructs containing the basic anti-XX-CAR scFv-CD8 hinge-41BB-CD3ζ in concert with ASS or OTC or ASS+OTC enzyme. A truncated CD34 is expressed for CAR-T identification and purification. (E) Western blot of ASS and OTC expression, with β-actin control, in control and modified anti-GD2-CAR-T cells posttransduction and expansion. (Representative of n = 7.) (F) ASS enzyme activity, measured by citrulline catabolism, is increased in ASS expressing CAR-T cells compared with controls. (G) OTC enzyme activity, measured by citrulline production, is increased in OTC expressing CAR-T cells compared with controls. (H) Highest proliferation of 2.5 × 106 modified anti-GD2-Jurkat CAR-T cell line following engraftment into NOG-SCID mice treated with recombinant human arginase (BCT-100, daily 50 mg/kg IV). (I) Proliferation of modified human CAR-T cells is significantly increased in low arginine media conditions, in vitro, as measured by flow cytometry after 72 hours. (J) Proliferation of modified anti-GD2 or anti-CD33-CAR-T cells is significantly increased in neuroblastoma (LAN-1 neuroblastoma or K562 AML, 72-hour conditioned supernatants) conditions, in vitro as measured by flow cytometry after 72 hours. (K) Expansion of modified CAR-T cells is enhanced in low arginine vs excess arginine (RPMI10%) conditions as measured by flow cytometry after 72 hours. (L) Liquid chromatography mass spectometry analysis of modified CAR-T cells after proliferation in low arginine conditions (72 hours) compared with each modified version. Heatmap demonstrating metabolomic changes in arginine and proline metabolism, pyrimidine metabolism, and purine metabolism pathways.
We hypothesized that insertion of ASS and/or OTC enzymes would allow CAR-T cells to refuel themselves, in a low arginine microenvironment. We reengineered previously described retroviral vectors containing CAR-41BB-3ζ expression cassettes against 4 common targets (anti-GD2, anti-CD33, anti-mesothelin, and anti-EGFRvIII; supplemental Figure 2B-I) to coexpress ASS, OTC, or both enzymes (Figure 1D; supplemental Figure 3A). CAR-T cells were expanded and sorted to a high degree of purity (supplemental Figure 3B). Western blots confirmed enhanced intracellular expression of ASS and OTC proteins compared with untransduced T cells and no enzyme controls (Figure 1E; supplemental Figure 3C-D). Next, we confirmed each enzyme insertion was metabolically active, catalyzing the increased conversion of citrulline to argininosuccinate (ASS; Figure 1F) or ornithine to citrulline (OTC) (Figure 1G).
To prove that the insertion of ASS and OTC leads to proliferative advantage, we first transduced a T-cell line with anti-GD2-ASS and anti -GD2-OTC (supplemental Figure 4A-B), and confirmed enzyme activity (supplemental Figure 4C-D). Enzyme additions resulted in increased CAR-T proliferation in vitro (supplemental Figure 4E). CAR-T cells were injected into arginine depleted mice to mimic tumor conditions. ASS- and OTC-modified CAR-T cells proliferated significantly compared with the GD2-CAR-T control (Figure 1H).
Next, we investigated the enzyme effects in CAR-T cells derived from multiple human donors, under low arginine culture conditions. The expression of ASS, OTC, or ASS+OTC enhanced CAR-T proliferation in vitro (Figure 1I; supplemental Figure 5A), regardless of scFv against CD33, GD2, mesothelin, or EGFRvIII (supplemental Figure 5B-E). Similar findings were identified in low arginine tumor cell-conditioned media (Figure 1J). Consistent with the metabolic adaptation, modified CAR-T proliferation is most enhanced under low arginine conditions, compared with when arginine is in excess (Figure 1K; supplemental Figure 5F-I). In contrast, untransduced T cells do not proliferate in the tumor microenvironment (supplemental Figure 6A-B).5,7 Under low arginine conditions, no improvement in proliferation is seen in the presence of ornithine or citrulline supplementation, corresponding to previous reports (supplemental Figure 6C).11-13
Analysis of PD-1, TIM3, and LAG3 immune checkpoint expression confirmed no significant changes in immune exhaustion (supplemental Figure 7A-B). No changes in cellular activation on target binding, as measured by interferon-γ, were observed (supplemental Figure 7C).To further investigate the broader impact of enzyme additions on downstream metabolites, CAR-T cells were subjected to untargeted metabolomics analysis by liquid chromatography-mass spectrometry.14 Each enzyme conferred a distinct metabolic phenotype. Nontransduced T cells are metabolically significantly different from CAR-T cells (supplemental Figure 8; supplemental Table 1).15 Analysis of proliferating enzyme-modified CAR-T cell metabolomes in low arginine conditions showed metabolic switching in arginine and proline metabolism, pyrimidine metabolism, and purine metabolism (Figure 1L; supplemental Table 2).
Our study strongly suggests that ASS or OTC improves CAR-T proliferation under tumor conditions. Extending our findings, we hypothesized that improving CAR-T proliferation would enhance control of hematological or solid cancer growth in vivo. We confirmed that insertion of enzymes led to no loss in cytotoxicity or specificity, using flow cytometry and chromium-51 release assays (Figure 2A-B; supplemental Figure 9A). No significant changes in degranulation or perforin release on recognition of target cells is seen, between the no-enzyme and modified CAR-T cells (supplemental Figure 9B). Modified anti-CD33 CAR-T cells were administered to AML cell line-engrafted mice. Expansion of CAR-T cells correlated with the frequency of AML in the bone marrow (Figure 2C). ASS-modified CAR-T cells had a significant peripheral expansion (Figure 2D; supplemental Figure 10A) and a corresponding reduction in AML frequency (Figure 2E; supplemental Figure 10B). We show that the ASS-CAR-T cells in the spleens of leukemia-bearing mice reexpand on restimulation with AML (supplemental Figure 10C).

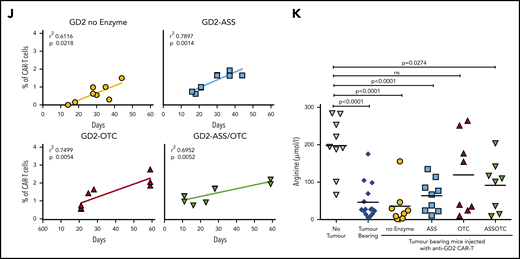
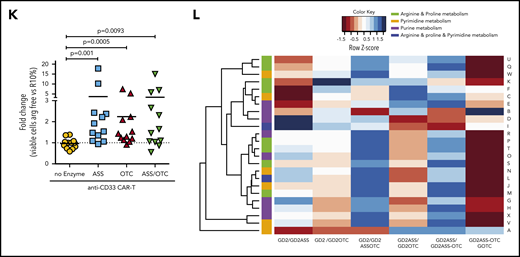
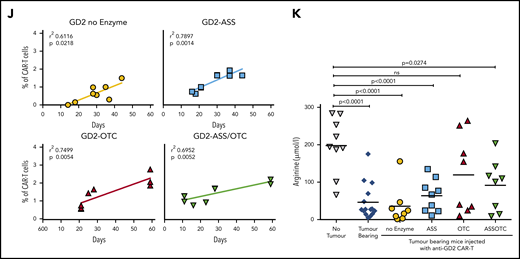
ASS and OTC enzymes increases CAR-T proliferation in vivo and activity against solid or hematological cancer. Specificity of modified CAR-T cell killing is unchanged by the addition of ASS or OTC enzymes, against (A) neuroblastoma or (B) AML target cells. Chromium (51Cr) release assays demonstrating antigen-specific killing of tumor cell targets by CAR-T cells. Minimal killing is seen against tumor cells that do not express the corresponding antigen. (C) Increased CAR-T proliferation correlates with a decreased AML burden in xenograft bone marrow. Linear regression line shown. (D) Increased proliferation of ASS-modified anti-CD33 CAR-T cells in NOG-SCID mice with established engraftment of AML blasts (HL60). (E) Decrease in established AML (HL60) engraftment in murine cell line xenografts treated with anti-CD33-ASS CAR-T cells. Data from day of euthanization. Dotted red line showing mean AML engraftment (43%) in the bone marrow of untreated mice. (F) Kaplan-Meier curves of GD2+ tumor murine xenografts, showing increased survival following treatment with CAR-T constructs. All remaining live mice were euthanized on day 57 for analysis. P value compared with control. (G) Increased proliferation of ASS, OTC, or ASS+OTC modified anti-GD2 CAR-T cells in nude mice with established s/c GD2+ tumors. Data from day of euthanization. (H) Anti-GD2-OTC CAR-T cells significantly decrease tumor growth in nude mice. (I) Kaplan-Meier curves of GD2+ tumor murine xenograft survival, which is increased with OTC-modified CAR-T constructs. All remaining live mice were euthanized on day 57 for analysis. P value compared with control. (J) Linear regression demonstrating the magnitude of anti-GD2-CAR-T cell proliferation correlates with GD2+ tumor xenograft survival. (K) Serum in tumor-bearing xenografts is deplete in arginine. Arginine concentrations are restored toward normal in response to CAR-T cell antitumor activity.
ASS and OTC enzymes increases CAR-T proliferation in vivo and activity against solid or hematological cancer. Specificity of modified CAR-T cell killing is unchanged by the addition of ASS or OTC enzymes, against (A) neuroblastoma or (B) AML target cells. Chromium (51Cr) release assays demonstrating antigen-specific killing of tumor cell targets by CAR-T cells. Minimal killing is seen against tumor cells that do not express the corresponding antigen. (C) Increased CAR-T proliferation correlates with a decreased AML burden in xenograft bone marrow. Linear regression line shown. (D) Increased proliferation of ASS-modified anti-CD33 CAR-T cells in NOG-SCID mice with established engraftment of AML blasts (HL60). (E) Decrease in established AML (HL60) engraftment in murine cell line xenografts treated with anti-CD33-ASS CAR-T cells. Data from day of euthanization. Dotted red line showing mean AML engraftment (43%) in the bone marrow of untreated mice. (F) Kaplan-Meier curves of GD2+ tumor murine xenografts, showing increased survival following treatment with CAR-T constructs. All remaining live mice were euthanized on day 57 for analysis. P value compared with control. (G) Increased proliferation of ASS, OTC, or ASS+OTC modified anti-GD2 CAR-T cells in nude mice with established s/c GD2+ tumors. Data from day of euthanization. (H) Anti-GD2-OTC CAR-T cells significantly decrease tumor growth in nude mice. (I) Kaplan-Meier curves of GD2+ tumor murine xenograft survival, which is increased with OTC-modified CAR-T constructs. All remaining live mice were euthanized on day 57 for analysis. P value compared with control. (J) Linear regression demonstrating the magnitude of anti-GD2-CAR-T cell proliferation correlates with GD2+ tumor xenograft survival. (K) Serum in tumor-bearing xenografts is deplete in arginine. Arginine concentrations are restored toward normal in response to CAR-T cell antitumor activity.
In solid tumors lentiviral- instead of retroviral-based manufacture of CAR-T cells is increasingly applied to generate CAR-T cells because of enhanced antitumor activity, which we reconfirmed in vivo (supplemental Figure 10D).16 Treatment of GD2+ tumor-bearing mice with anti-GD2 CAR-T cells significantly improved overall survival (Figure 2F). Enzyme-modified CAR-T cell numbers were increased in the peripheral blood (Figure 2G). OTC-CAR-T cells led to a significant reduction in tumor growth rate (Figure 2H; supplemental Figure 11A) and prolonged murine survival (Figure 2I). Modified CAR-T proliferation correlated with murine survival (Figure 2J) and restored plasma arginine levels to normal (Figure 2K).
Amino acid uptake by tumor cells through CAT-1 or LAT-1 will limit the availability of microenvironmental ornithine or citrulline respectively, for CAR-T cells.11,17 Quantitative polymerase chain reaction of sorted tumor cells from murine xenografts identified relatively high expression of CAT-1 on bone marrow-derived AML cells but higher LAT-1 on neuroblastoma cells (supplemental Figure 11B-C).18,19 Therefore, ASS expression in CAR-T cells may provide an advantage in AML by catabolizing citrulline as a substrate instead, whereas OTC expression in CAR-T cells may be of benefit in neuroblastoma by allowing catabolism of residual ornithine. These findings are consistent with the response of ASS- or OTC-modified CAR-T cells in our 2 models. Profiling of the amino acid requirements of adult and pediatric tumors could allow for rationale selection of enzyme-modified CAR-T cells in clinical trials.
Although CAR-T cells have been shown to induce tumor cell death, optimal activity may be limited by a number of causes including low target antigen burden, poor penetrance into the tumor microenvironment, or a lack of CAR-T cell potency to sustain T-cell expansion and persistence.20,21 Strategies that have attempted to boost CAR-T activity by altering costimulation, enhancing conditioning regimens, or through manipulation of microenvironment cytokines have not led to dramatic clinical improvements.22,23 Altered metabolism is one of the core pillars of cancer biology, yet approaches to tackle the immunosuppressive microenvironment that drains the fuel to drive CAR-T proliferation, persistence and thus activity have been limited. Attempts to inhibit tumor or myeloid cell amino acid catabolizing enzymes with arginase and IDO inhibitors had only very limited efficacy in humans.24,25 Supplementing patients with amino acid substrates could also be attempted, but here there is a major risk of increasing tumor growth and metastases. Here, we suggest the first use of enzyme expression as a technologically novel solution to adapt CAR-T cells to their metabolic environment and thus enhance antitumor activity. Reengineering T cells to express metabolic enzymes could also be widely applicable to other forms of T-cell therapy. We propose to take these findings forward into a phase 1/2 clinical trial.
For original data, please contact francis.mussai@nhs.net.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and parents who contributed samples to the study. The authors also thank Janet Morse, Jane Cooper, Cay Shakespeare, and Sarah-Jane Staveley for consenting of patients and collection of samples for this study.
This work was supported by Cancer Research UK, Treating Children with Cancer, Amber Phillpott Trust, Birmingham Children’s Hospital, and the alumni and donors to the University of Birmingham. This work was supported by Phenome Centre Birmingham (MR/M009157/1).
Authorship
Contribution: F.M. and C.D.S. designed the study, supervised research, analyzed data, secured funding, and wrote the manuscript; F.M. additionally secured ethical approval and was chief investigator of the study; L.F. designed and performed research; S.B., O.Y., B.M.d.C., V.T., S.P., and V.N. performed research; S.P.L. supervised research; L.C. supervised murine research and secured funding and ethical approval; A.J., G.L., A.S., and W.B.D. acquired and interpreted the metabolomics data; and all authors contributed to writing of the manuscript.
Conflict-of-interest disclosure: F.M. and C.D.S. are listed as inventors on the following patent: PCT/GB2018/053771 (published as WO2019/122936) pertaining to this study. S.P.L. is listed as an inventor on the following patent application/patent families: CT/GB2017/050686 (published as WO2017/158337) and PCT/GB2017/050689 (WO2017/158339), and has interests in anti-CLEC14A CAR-T cells, studies relating to which have been supported by Cell and Gene Therapy Catapult UK and licensed to Chimeric Therapeutics Ltd. The remaining authors declare no competing financial interests.
Correspondence: Francis Mussai, Institute of Immunology and Immunotherapy, University of Birmingham, Birmingham, B15 2TT, United Kingdom; e-mail: francis.mussai@nhs.net.