Key Points
TanCAR7 T cells showed dual antigen targeting and formed superior IS structures, which may be related to their robust antitumor activity.
TanCAR7 T cells elicited a potent and durable antitumor response but not grade 3 or higher CRES in patients with r/rNHL.

Abstract
Chimeric antigen receptor (CAR) T cells targeting CD19 have achieved breakthroughs in the treatment of hematological malignancies, such as relapsed/refractory non-Hodgkin lymphoma (r/rNHL); however, high rates of treatment failure and recurrence after CAR T-cell therapy are considerable obstacles to overcome. In this study, we designed a series of tandem CARs (TanCARs) and found that TanCAR7 T cells showed dual antigen targeting of CD19 and CD20, as well as formed superior and stable immunological synapse (IS) structures, which may be related to their robust antitumor activity. In an open-label single-arm phase 1/2a trial (NCT03097770), we enrolled 33 patients with r/rNHL; 28 patients received an infusion after conditioning chemotherapy. The primary objective was to evaluate the safety and tolerability of TanCAR7 T cells. Efficacy, progression-free survival, and overall survival were evaluated as secondary objectives. Cytokine release syndrome occurred in 14 patients (50%): 36% had grade 1 or 2 and 14% had grade 3. No cases of CAR T-cell–related encephalopathy syndrome (CRES) of grade 3 or higher were confirmed in any patient. One patient died from a treatment-associated severe pulmonary infection. The overall response rate was 79% (95% confidence interval [CI], 60-92%), and the complete response rate was 71%. The progression-free survival rate at 12 months was 64% (95% CI, 43-79%). In this study, TanCAR7 T cells elicited a potent and durable antitumor response, but not grade 3 or higher CRES, in patients with r/rNHL.
Introduction
Chimeric antigen receptors (CARs) are synthetic receptors for antigens that reprogram T-cell specificity, function, and persistence.1 T cells engineered with a CD19-targeting CAR exhibit remarkable efficacy in patients with hematological malignancies, such as B-cell acute lymphocytic leukemia (B-ALL)2-4 and B-cell lymphoma.5-7 Despite this impressive efficacy, progressive disease (PD) occurs in a large proportion of patients who receive a CAR T-cell infusion,8 primarily as a result of a lack of CAR T-cell persistence and tumor cell resistance stemming from antigen loss or reduced antigen expression below the threshold required for CAR T-cell activity.9-11 Sotillo and colleagues have described in detail the escape mechanisms associated with antigen loss in B-ALL during CD19 CAR T-cell therapy; these mechanisms include alternative splicing of CD19, frameshift mutations, and missense mutations.10 In addition, a recent study demonstrated that CAR T cells transfer target antigens on the tumor cell surface to their own surface by trogocytosis, decreasing the density of target antigens on tumor cells and suppressing T-cell activity by promoting T-cell killing and exhaustion.12 Unlike the case for B-ALL patients, biopsies are not always obtained from non-Hodgkin lymphoma (NHL) patients at the time of relapse, so the incidence of CD19− relapse remains less clear; however, emerging data provide evidence that this phenomenon also occurs in NHL.5,7
Multiple studies have shown that simultaneously targeting 2 antigens with CAR T cells may reduce the likelihood of antigen escape by tumor cells and potentially increase tumor cell–killing activity.8,12-14 Grada and colleagues reported a single-chain bispecific CAR targeting CD19 and human epidermal growth factor receptor 2 (HER2).15 This bispecific receptor, called tandem CAR (TanCAR), efficiently triggered T-cell activation in response to CD19 or HER2. Although TanCAR remains a proof-of-concept of Boolean “OR”-gated signal computation, because both antigens are typically not expressed by the same cell, these findings fueled our interest in TanCAR targeting of CD19 and CD20 to overcome antigen escape-mediated relapse after CD19- or CD20-directed therapy.
Here, we designed a series of TanCARs targeting CD19 and CD20 and found that TanCAR7 T cells show dual antigen coverage and elicit a potent and durable antitumor response. Furthermore, we conducted an open-label single-arm phase 1/2a trial to explore the safety and tolerability of TanCAR7 T cells in patients with relapsed/refractory non-Hodgkin lymphoma (r/rNHL).
Methods
Trial design
A single-arm phase 1/2a clinical trial (NCT03097770) was designed to evaluate the safety, efficacy, and feasibility of administering autologous TanCAR7 T cells to patients with relapsed or refractory B-cell lymphoma. This study was approved by the Ethics Committee of the Chinese PLA General Hospital, and informed consent was obtained from all patients. Patients were recruited and treated at the Chinese PLA General Hospital. Two or 3 days before the infusion, all patients received lymphodepleting doses of cyclophosphamide (20-30 mg/kg divided over 3 days) and fludarabine (20-30 mg/m2 × 3 days), with or without doxorubicin liposome (10 mg/m2 on 1 day).
Efficacy evaluations were based on recommendations by the International Conference on Malignant Lymphomas Imaging Working Group (Cheson Response Criteria and The Lugano Classification 2014); additional assessments were included for chronic lymphocytic leukemia (CLL)/small lymphocytic leukemia (SLL) patients based on the consensus guidelines of the International Workshop on CLL.16,17 An Independent Review Committee, appointed by the PLA General Hospital, reviewed the data related to disease-response assessments according to the Guideline for Efficacy Evaluation in NHL Studies. Cytokine release syndrome (CRS) was assessed and graded according to the CRS grading system developed by Lee et al.18 Neurological toxicity was assessed and graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE), version 4.0.
The investigator was responsible for ensuring that all adverse events (AEs) observed directly or reported by the subject between enrollment (ie, commencement of leukapheresis) and 3 months after TanCAR7 T-cell infusion were monitored and reported. After 3 months, target AEs, including neurological events, hematological events, infections, autoimmune disorders, and secondary malignancies, were monitored and reported for 24 months after treatment with TanCAR7 T cells or until PD was observed. CTCAE, version 4.0 was used to grade AEs. Further details regarding the study procedures are provided in supplemental Methods (available on the Blood Web site).
CAR construct and CAR T-cell manufacturing
A CD19 CAR, CD20 CAR, and TanCAR1-8 were generated by linking the CD19 single-chain variable fragment (scFv) derived from the FMC63 monoclonal antibody19 or the CD20 scFv derived from the Leu-16 monoclonal antibody20 in frame with the hinge and transmembrane domains of CD8 and the cytoplasmic domains of 4-1BB and CD3 ζ. A schematic diagram of the CAR constructs is shown in Figure 1A, and the scFv sequence of TanCAR7 is provided in supplemental Figure 1. The preparation and quality control of CAR T cells are described in supplemental Methods.
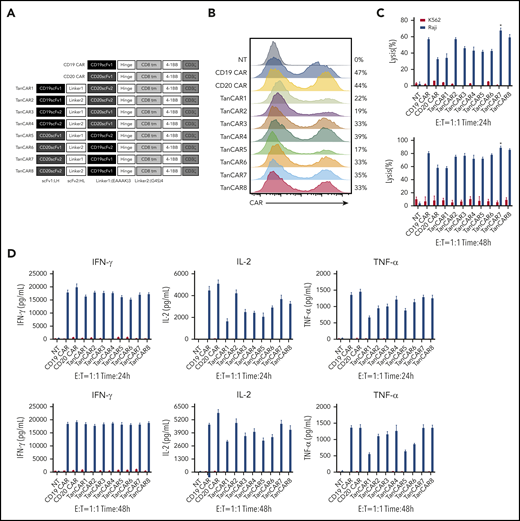
Development of tandem CARs. (A) Schematic diagram of the recombinant lentiviral vectors encoding the prototype anti-CD19 CAR (CD19 CAR), anti-CD20 CAR (CD20 CAR), and TanCARs generated in this study. (B) Flow cytometry analysis showing the expression levels of CARs encoded by the indicated constructs. Graphs are representative of 3 independent experiments. (C) Cytotoxic activity of TanCAR, CD19 CAR, and CD20 CAR T cells using 24-hour or 48-hour CytoTox 96 Non-Radioactive Cytotoxicity Assays with Raji cells or K562 cells at an E:T ratio of 1:1. *P < .05, TanCAR7 vs CD19 CAR/TanCAR8; P < .01, TanCAR7 vs TanCAR2/TanCAR4; P < .001, TanCAR7 vs TanCAR1/TanCAR3/TanCAR5/TanCAR6; P < .0001, TanCAR7 vs CD20 CAR (upper panel). *P < .05, TanCAR7 vs CD19 CAR/TanCAR3; P < .01, TanCAR7 vs TanCAR2/TanCAR4/TanCAR5/TanCAR6; P < .001, TanCAR7 vs CD20 CAR/TanCAR1 (lower panel). (D) Cytokine production by CD19 CAR, CD20 CAR, and TanCAR7 T cells cocultured with Raji or K562 cells at an E:T ratio of 1:1, as measured by enzyme-linked immunosorbent assay. All data are mean ± standard deviation. Experiments were repeated with 3 pools of donor-derived T cells (n = 3). 2-tailed unpaired 2-sample Student t test. NT, control nontransduced T cells. IFN-γ, interferon-γ; NT, nontransduced; TNF-α, tumor necrosis factor-α.
Development of tandem CARs. (A) Schematic diagram of the recombinant lentiviral vectors encoding the prototype anti-CD19 CAR (CD19 CAR), anti-CD20 CAR (CD20 CAR), and TanCARs generated in this study. (B) Flow cytometry analysis showing the expression levels of CARs encoded by the indicated constructs. Graphs are representative of 3 independent experiments. (C) Cytotoxic activity of TanCAR, CD19 CAR, and CD20 CAR T cells using 24-hour or 48-hour CytoTox 96 Non-Radioactive Cytotoxicity Assays with Raji cells or K562 cells at an E:T ratio of 1:1. *P < .05, TanCAR7 vs CD19 CAR/TanCAR8; P < .01, TanCAR7 vs TanCAR2/TanCAR4; P < .001, TanCAR7 vs TanCAR1/TanCAR3/TanCAR5/TanCAR6; P < .0001, TanCAR7 vs CD20 CAR (upper panel). *P < .05, TanCAR7 vs CD19 CAR/TanCAR3; P < .01, TanCAR7 vs TanCAR2/TanCAR4/TanCAR5/TanCAR6; P < .001, TanCAR7 vs CD20 CAR/TanCAR1 (lower panel). (D) Cytokine production by CD19 CAR, CD20 CAR, and TanCAR7 T cells cocultured with Raji or K562 cells at an E:T ratio of 1:1, as measured by enzyme-linked immunosorbent assay. All data are mean ± standard deviation. Experiments were repeated with 3 pools of donor-derived T cells (n = 3). 2-tailed unpaired 2-sample Student t test. NT, control nontransduced T cells. IFN-γ, interferon-γ; NT, nontransduced; TNF-α, tumor necrosis factor-α.
Laboratory assays
Experimental samples were analyzed by flow cytometry, enzyme-linked immunosorbent assay, immunofluorescence, quantitative polymerase chain reaction (qPCR), RNA sequencing, and RNA in situ hybridization (ISH). Assay details are provided in supplemental Methods.
Statistical analysis
All statistical analyses were performed using Prism 7 (GraphPad) software. Comparisons of 2 groups were performed by 2-tailed parametric or nonparametric (Kolmogorov-Smirnov test) Student t tests for unpaired data. A log-rank Mantel-Cox test was used to compare survival differences between groups. P values <.05 were considered statistically significant. The detailed statistical plan for the clinical trial is described in supplemental Methods.
Results
Development of tandem CARs
We designed a series of single-chain bispecific CARs by optimizing the order of anti-CD19 and anti-CD20 scFv’s (constructed as VHVL-VLVH or VLVH-VHVL to avoid the formation of an artificial scFv between the 2 scFv’s) and the type of linker between the scFv’s; we called these CARs TanCAR1-8 (Figure 1A). All CARs were differentially expressed in lentivirus-transduced human peripheral blood T cells (Figure 1B). Notably, despite the reduced production of cytokines, such as interferon-γ, tumor necrosis factor-α, and interleukin-2 (IL-2), TanCAR7 T cells exhibited more potent antitumor activity than single-targeted CAR T cells (CD19 CAR and CD20 CAR) or other TanCAR T cells when they interacted with CD19+CD20+ Raji lymphoma cells (Figures 1C-D, 2F).

TanCAR7 T cells formed a stable IS and showed faster degranulation than CD19 CAR or CD20 CAR T cells. (A) Representative confocal imaging of synapses formed by CAR T cells. CD19 CAR, CD20 CAR, and TanCAR7 T cells were loaded onto CD19- and CD20-coated lipid bilayers for 10 minutes, fixed, and stained for CAR (green) and F-actin (red). Scale bars, 2 μm. (B) Quantification of IS structures along the lipid bilayer–T-cell focal plane was performed by measuring the synapse area and the mean fluorescence intensity (MFI) of F-actin at 10, 20, and 30 minutes. Three independent experiments were performed, and representative results are shown. Data are mean ± standard deviation (SD). For each group, 7 to 39 random CAR+ cells were imaged and evaluated. P values were calculated with the Kolmogorov-Smirnov test. (C) Representative MTOC confocal imaging of CAR T cells (left panels). Sorted CD19 CAR, CD20 CAR, and TanCAR7 T cells were loaded onto CD19- and CD20-coated lipid bilayers for 10 minutes. Fixed and permeabilized CAR T cells were stained for γ tubulin (green), F-actin (red), and the nucleus (Hoechst 33342, blue). MTOC polarization at the IS. Data are mean ± SD (right panel). For each group, 20 to 30 random cells were imaged and evaluated. Three independent experiments were performed, and representative results are shown. P values were calculated with the Kolmogorov-Smirnov test. Scale bars, 2 μm. (D) CAR T cells assessed by flow cytometry. The cells were labeled with Fluo-4, AM, stimulated with Raji cells or phorbol 12-myristate-13-acetate/ionomycin, and evaluated by flow cytometry. Three independent experiments were performed, and representative results are shown. CD107a (E) and cytokine (F) expression by CAR T cells, as determined by the percentage of positive cells in flow cytometric analyses. Three independent experiments were performed, and representative results are shown. All data are mean ± SD. *P < .05, **P < .01, ***P < .001, ****P < .0001. IFN-γ, interferon-γ; ns, not significant (P > .05); NT, nontransduced; PMA, phorbol 12-myristate-13-acetate; TNF-α, tumor necrosis factor-α.
TanCAR7 T cells formed a stable IS and showed faster degranulation than CD19 CAR or CD20 CAR T cells. (A) Representative confocal imaging of synapses formed by CAR T cells. CD19 CAR, CD20 CAR, and TanCAR7 T cells were loaded onto CD19- and CD20-coated lipid bilayers for 10 minutes, fixed, and stained for CAR (green) and F-actin (red). Scale bars, 2 μm. (B) Quantification of IS structures along the lipid bilayer–T-cell focal plane was performed by measuring the synapse area and the mean fluorescence intensity (MFI) of F-actin at 10, 20, and 30 minutes. Three independent experiments were performed, and representative results are shown. Data are mean ± standard deviation (SD). For each group, 7 to 39 random CAR+ cells were imaged and evaluated. P values were calculated with the Kolmogorov-Smirnov test. (C) Representative MTOC confocal imaging of CAR T cells (left panels). Sorted CD19 CAR, CD20 CAR, and TanCAR7 T cells were loaded onto CD19- and CD20-coated lipid bilayers for 10 minutes. Fixed and permeabilized CAR T cells were stained for γ tubulin (green), F-actin (red), and the nucleus (Hoechst 33342, blue). MTOC polarization at the IS. Data are mean ± SD (right panel). For each group, 20 to 30 random cells were imaged and evaluated. Three independent experiments were performed, and representative results are shown. P values were calculated with the Kolmogorov-Smirnov test. Scale bars, 2 μm. (D) CAR T cells assessed by flow cytometry. The cells were labeled with Fluo-4, AM, stimulated with Raji cells or phorbol 12-myristate-13-acetate/ionomycin, and evaluated by flow cytometry. Three independent experiments were performed, and representative results are shown. CD107a (E) and cytokine (F) expression by CAR T cells, as determined by the percentage of positive cells in flow cytometric analyses. Three independent experiments were performed, and representative results are shown. All data are mean ± SD. *P < .05, **P < .01, ***P < .001, ****P < .0001. IFN-γ, interferon-γ; ns, not significant (P > .05); NT, nontransduced; PMA, phorbol 12-myristate-13-acetate; TNF-α, tumor necrosis factor-α.
Analysis of the phenotype and function of TanCAR7 T cells compared with single-targeted CAR T cells
To evaluate the single-antigen recognition ability of TanCAR7, CRISPR-Cas9 gene editing was used to delete CD19 or CD20 from Raji cells (supplemental Figure 2A). As expected, CD19 CAR or CD20 CAR T cells showed no response to the corresponding mutant Raji cells (RajiCD19KO and RajiCD20KO). In contrast, TanCAR7 T cells efficiently killed wild-type and mutant Raji cells (supplemental Figure 2B-C). Remarkably, TanCAR7 T cells exhibited higher antitumor activity than did single-targeted CAR T cells at a low effector-to-target (E:T) ratio (supplemental Figure 2D). All CAR T cells expressed similar amounts of CD62L, CD45RO, PD-1, and TIM-3 when cultured without CD19 or CD20 antigen stimulation (supplemental Figure 3A); however, the TanCAR7 T-cell population contained a higher percentage of CD62L+CD45RO+ central memory T cells and a lower percentage of differentiated CD62L−CD45RO+ effector cells upon interaction with Raji cells (supplemental Figure 3B). PD-1 and TIM-3 expression was also lower in TanCAR7 T cells than in single-targeted CAR T cells upon antigen exposure (supplemental Figure 3C). All CAR T cells exhibited equivalent proliferation upon interaction with Raji cells (supplemental Figure 3D-E).
TanCAR7 T cells exhibit a stable IS and faster degranulation than single-targeted CAR T cells
To investigate the functional basis for the major differences in antitumor efficacy between TanCAR7 and single-targeted CAR T cells, we further examined the responses induced by these CARs. The immunological synapse (IS), a discrete structural entity, forms after the ligation of specific activating receptors and leads to the destruction of tumor cells.21,22 The placement of different scFv domains in a CAR-formed IS may result in different antitumor activities.13,23 In previous studies, F-actin accumulation at the IS and polarization of the microtubule organizing center (MTOC) were measured to assess whether CAR T cells have a functional advantage.24,25 The inhibition of IS formation by an F-actin or Lck inhibitor blocked CAR T-cell activation and tumor cell killing (supplemental Figure 4). Compared with single-targeted CAR T cells, TanCAR7 T cells showed significantly higher levels of MTOC polarization to the IS, as indicated by a reduced distance between the MTOC and IS, as well as significantly higher F-actin accumulation at the IS, which produced a superior and more stable IS structure (Figure 2A-C). In addition, the inhibition of IS formation by an F-actin inhibitor or Lck inhibitor blocked CAR T-cell activation and killing activity on tumor cells (supplemental Figure 4). Furthermore, we found that, although all CAR T cells exhibited similar calcium influx following stimulation with phorbol 12-myristate-13-acetate/ionomycin, TanCAR7 T cells exhibited greater calcium influx than did single-targeted CAR T cells while interacting with Raji cells (Figure 2D). In general, an increase in intracellular calcium levels triggers the degranulation machinery and the translocation of granules from the cell interior toward the plasma membrane in a microtubule-dependent manner.26,27 To determine whether increased calcium influx influences the intensity of degranulation, we next assessed degranulation in TanCAR7 and single-targeted CAR T cells following interaction with Raji cells. As expected, TanCAR7 T cells exhibited stronger and more rapid degranulation than single-targeted CAR T cells (Figure 2E). The secretion of intracellular factors was consistent with the results obtained for cytokines in the supernatant (Figure 2F).
Transcriptional profiles of CD19 CAR, CD20 CAR, and TanCAR7 T cells exposed to antigen
To further characterize these different structural, phenotypic, and functional patterns, we compared the genome-wide transcriptional profiles of CD19 CAR, CD20 CAR, and TanCAR7 T cells following interaction with Raji cells. Consistent with the functional studies, gene enrichment analysis revealed major differences in transcriptomic profiles related to T-cell activation/exhaustion and cytokine production, as well as in profiles related to IS structure and activity (supplemental Figure 5A). As expected, TanCAR7 T cells showed greater upregulation of gene expression profiles related to calcium flux and IS structure and activity (supplemental Figure 5B). The upregulation of the expression of key genes involved in calcium signaling, such as ITPR3 and PKD1,28,29 and the concurrent increase in the expression of IS activity–associated genes, such as CCDC88B and PTK2,30,31 corroborated the rapid degranulation seen in TanCAR7 T cells (supplemental Figure 5B). Moreover, gene expression analysis showed that the expression levels of gene sets associated with cytokine activity and production were significantly lower in TanCAR7 T cells than in CD20 CAR T cells (supplemental Figure 5B). We also found that CD20 CAR T cells exhibited significantly higher expression levels of T-cell activation/exhaustion-related genes than compared with TanCAR7 and CD19 CAR T cells. Despite exhibiting similar expression levels, TanCAR7 T cells expressed lower levels of key genes, such as CTLA4 and TIGIT,32-34 than were found in CD19 CAR T cells (supplemental Figure 5B). Collectively, these findings underscore the critical roles of different antigen-antibody binding types in influencing T-cell fate.
TanCAR7 T cells showed superior antitumor activity in vivo
Next, we compared the antitumor activity of TanCAR7 T cells and single-targeted CAR T cells in vivo using a lymphoma xenograft model in NSG mice. Raji-luciferase-GFP cells (1 × 106) were injected IV via the tail vein. Compared with CD19 CAR T cells, CD20 CAR T cells showed markedly lower antitumor activity and achieved only a transient reduction in tumor burden, whereas the activity of TanCAR7 T cells exceeded that of CD19 CAR T cells, with rapid and strong CAR T-cell expansion and the induction of long-term remission in all mice (supplemental Figure 6A-D). In addition, TanCAR7 T cells in the bone marrow expressed relatively low levels of PD-1 and TIM-3 (supplemental Figure 6E), consistent with the in vitro results. Furthermore, we observed that, in a solid tumor xenograft mouse model inoculated intraperitoneally, TanCAR7 T cells showed antitumor effects superior to those of CD19 CAR or CD20 CAR T cells (Figure 3). RNA ISH showed that TanCAR7 T cells exhibited better intratumoral infiltration and higher granzyme B levels than CD19 CAR and CD20 CAR T cells (Figure 3F). Taken together, these data indicate that the antitumor potential of TanCAR7 T cells is superior to that of single-targeted CAR T cells.

TanCAR7 T cells showed superior antisolid tumor activity in vivo. NSG mice (n = 6) were injected intraperitoneally with 2 × 106 Raji-luciferase-GFP cells mixed with Matrigel on day −7 to establish a solid tumor xenograft animal model. On day 0, the mice were injected IV with 1 × 106 CAR T cells. (A) The bioluminescence images (BLIs) indicating the mouse tumor burden at the indicated time points are representative of all experiments (n = 6 mice per group; the results were pooled from 2 independent experiments). (B) Mouse tumor burden (average radiance). (C) The percentage of CD3+ T cells in peripheral blood was used to evaluate the expansion of CAR T cells. Data for each mouse at the indicated time points are presented. (D) Survival analyses of mice treated with TanCAR7 T cells, NT cells, CD19 CAR T cells, or CD20 CAR T cells. P values were calculated with the log-rank Mantel-Cox test. (E-F) Mice were euthanized on day 7 after treatment. (E) Flow cytometry was used to determine the percentage of PD-1+TIM-3+ CAR T cells in peritoneal tumor tissue. (F) Paraffin tumor sections were stained with an scFv-specific probe or a granzyme B–specific probe via RNA ISH (original magnification ×100 and ×400) (left panel). The positive rates of scFV and Granzyme B are shown on the right. Data are mean ± standard deviation of 5 mice per group in 2 independent experiments. P values were calculated using a 2-tailed unpaired Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001. IFN-γ, interferon-γ; NT, nontransduced control; TNF-α, tumor necrosis factor-α.
TanCAR7 T cells showed superior antisolid tumor activity in vivo. NSG mice (n = 6) were injected intraperitoneally with 2 × 106 Raji-luciferase-GFP cells mixed with Matrigel on day −7 to establish a solid tumor xenograft animal model. On day 0, the mice were injected IV with 1 × 106 CAR T cells. (A) The bioluminescence images (BLIs) indicating the mouse tumor burden at the indicated time points are representative of all experiments (n = 6 mice per group; the results were pooled from 2 independent experiments). (B) Mouse tumor burden (average radiance). (C) The percentage of CD3+ T cells in peripheral blood was used to evaluate the expansion of CAR T cells. Data for each mouse at the indicated time points are presented. (D) Survival analyses of mice treated with TanCAR7 T cells, NT cells, CD19 CAR T cells, or CD20 CAR T cells. P values were calculated with the log-rank Mantel-Cox test. (E-F) Mice were euthanized on day 7 after treatment. (E) Flow cytometry was used to determine the percentage of PD-1+TIM-3+ CAR T cells in peritoneal tumor tissue. (F) Paraffin tumor sections were stained with an scFv-specific probe or a granzyme B–specific probe via RNA ISH (original magnification ×100 and ×400) (left panel). The positive rates of scFV and Granzyme B are shown on the right. Data are mean ± standard deviation of 5 mice per group in 2 independent experiments. P values were calculated using a 2-tailed unpaired Student t test. *P < .05, **P < .01, ***P < .001, ****P < .0001. IFN-γ, interferon-γ; NT, nontransduced control; TNF-α, tumor necrosis factor-α.
Baseline characteristics of patients and TanCAR7 T-cell infusion
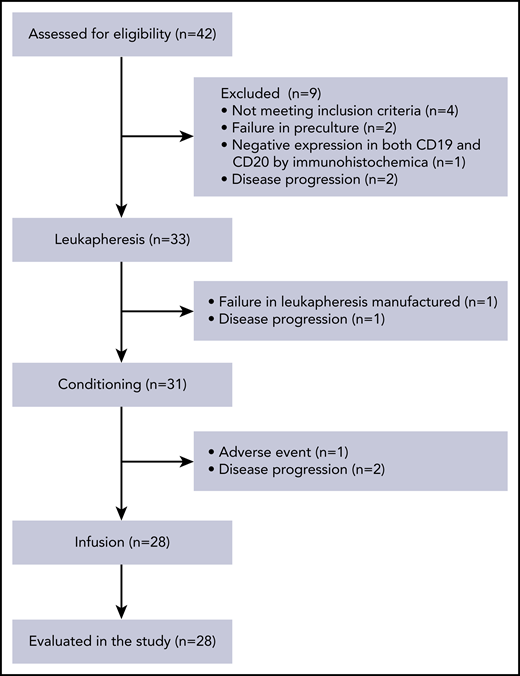
We performed a phase 1/2a trial of TanCAR7 T-cell therapy in patients with NHL who had refractory disease, despite undergoing the recommended prior therapy. Thirty-three patients were enrolled between May of 2017 and September of 2018. TanCAR7 T cells were successfully manufactured for 31 patients: 1 patient failed leukapheresis, and another had AEs after leukapheresis. The manufactured TanCAR7 T cells were administered to 28 patients. Before infusion, 1 patient had serious AEs, and 2 could not tolerate pretreatment because of disease progression (Figure 4). The median number of days from enrollment to infusion was 26 (range, 22-29). All patients who received TanCAR7 T cells were included in the AE analysis, and 27 were included in the efficacy evaluation. The data cutoff date for the efficacy evaluation was February of 2020, resulting in a median follow-up of 19.1 months. The patient characteristics are shown in Table 1 and supplemental Tables 1 through 3. No bridging chemotherapy regimen was administered after the last treatment prior to enrollment in our clinical trial. After a conditioning regimen of cyclophosphamide and fludarabine, the first 7 patients received infusions in the range of 0.5 × 106 to 6 × 106 cells per kilogram of body weight (supplemental Tables 1 and 4). Efficacy was evaluated based on the recommendations of the International Conference on Malignant Lymphomas Imaging Working Group at 3, 6, 12, 18, 24, and 36 months postinfusion. Six patients had an objective response, and 5 achieved a complete response (CR). Three patients experienced grade ≤ 2 cytokine release syndrome (CRS), and no serious AEs occurred (Figure 5; supplemental Table 1). The results demonstrated that infusion at a dose of 0.5 × 106 to 6 × 106 TanCAR7 T cells per kilogram is safe and has manageable toxicity. Subsequently, an extended dose range of cell infusion was tested; 21 r/rNHL patients were treated with TanCAR7 T cells at a target dose of 1 × 106 to 8 × 106 cells per kilogram after receiving a conditioning regimen (supplemental Tables 1 and 4). To reduce the tumor burden before infusion and, thereby, decrease the risk of CRS,35,36 fludarabine/cyclophosphamide plus doxorubicin was administered to patients as specified in supplemental Table 1.
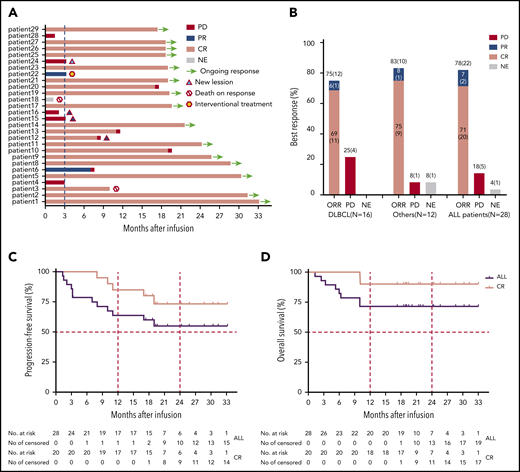
Clinical efficacy of TanCAR7 T cells in patients with r/rNHL. (A) Durations of the response and survival after the infusion of TanCAR7 T cells. Patient 3 died of severe pneumonia outside of our institute after achieving a CR 10 months postinfusion, and patient 18 died from a severe pulmonary infection associated with persistent unrecoverable myelosuppression. Four patients had new lesions in the context of controlled primary lesions. (B) Objective response rate (ORR) among the 28 treated patients; it was calculated by dividing the number of patients with a CR or PR by the number of the patients who received TanCAR7 T cells. Kaplan-Meier estimates of progression-free survival (C) and overall survival (D). The rates were calculated using data for all 28 patients who received an infusion. ALL, acute lymphocytic leukemia; NE, not evaluated; PR, partial response.
Clinical efficacy of TanCAR7 T cells in patients with r/rNHL. (A) Durations of the response and survival after the infusion of TanCAR7 T cells. Patient 3 died of severe pneumonia outside of our institute after achieving a CR 10 months postinfusion, and patient 18 died from a severe pulmonary infection associated with persistent unrecoverable myelosuppression. Four patients had new lesions in the context of controlled primary lesions. (B) Objective response rate (ORR) among the 28 treated patients; it was calculated by dividing the number of patients with a CR or PR by the number of the patients who received TanCAR7 T cells. Kaplan-Meier estimates of progression-free survival (C) and overall survival (D). The rates were calculated using data for all 28 patients who received an infusion. ALL, acute lymphocytic leukemia; NE, not evaluated; PR, partial response.
Outcomes in r/rNHL patients
In a total of 28 patients, the best overall response rate was 79% (95% confidence interval [CI], 60-92%), 71% of the patients achieved a CR (95% CI, 51-87%), and 7% had a partial response (PR) (95% CI, 1-24%) (Figure 5A-B; supplemental Figure 7). Of the 4 patients who were CD19− at baseline by flow cytometry and immunohistochemistry, 2 achieved a CR as the best response, 1 had a PR, and 1 showed PD. Whether TanCAR7 is less effective in such patients should be assessed in more populations. The median progression-free survival was not reached for all of the treated patients; the progression-free survival rates were 79% (95% CI, 58-90%) at 6 months and 64% (95% CI, 43-79%) at 12 months (Figure 5C). Among patients who had a CR at 3 months, estimated progression-free survival was 85% (95% CI, 60-95%) at 12 months (Figure 5C). Among patients with diffuse large B-cell lymphoma, 12 of 16 (75%) had a response (95% CI, 48-93%), the median progression-free survival was not reached, and 75% (95% CI, 46-90%) showed no disease progression at 12 months after infusion (supplemental Figure 8). In an intent-to-treat analysis that included all 33 enrolled patients, the overall response rate was 67% (95% CI, 48-82%), and the estimated probability of progression-free survival at 12 months was 59% (95% CI, 40-75%) (supplemental Figure 9). The median event-free survival was not reached, with an estimated rate of 61% (95% CI, 40-76%) at 12 months (supplemental Figure 10). The median overall survival was not reached, with overall survival rates of 82% (95% CI, 62-92%) at 6 months and 71% (95% CI, 51-85%) at 12 months. Among the patients who had a CR at 3 months, the estimated overall survival rate was 90% (95% CI, 66-97%) at 12 months (Figure 5D).
At the time of data cutoff, 10 patients had PD, including 6 with diffuse large B-cell lymphoma, 2 with transformed follicular lymphoma, 1 with follicular lymphoma, and 1 with CLL/SLL. Among these patients, 9 had stage IV disease, 5 had an Eastern Cooperative Oncology Group performance status score of 2, 4 had bulky disease, 8 were positive for Ki-67 in >70% of lesions at baseline, and 3 had PD after autologous hematopoietic stem cell transplantation (Figure 5A; supplemental Tables 1 and 2). Three other patients started alternative therapy before disease progression (n = 1) or died of severe pneumonia inside (n = 1) or outside of (n = 1) our institute. Fifteen patients remained in remission. Four patients relapsed after TanCAR7 T-cell infusion, 1 because of antigen loss, as determined by immunohistochemistry of a biopsy (supplemental Figure 11).
Adverse events
AEs of special interest that may be related to TanCAR7 T-cell therapy are shown in Table 2. The most common grade 3 or higher events observed within 1 month of infusion included neutropenia (61%), pyrexia (36%), and decreased platelet count (25%). CRS occurred in 14 patients (50%), 29% with grade 1 AEs, 6% with grade 2 AEs, and 14% with grade 3 AEs. The median time from infusion to the onset of CRS was 1 day (range, 1-5), whereas the median duration of CRS was 6 days (range, 1-9). The median time to the onset of grade 3 CRS was 1 day (range, 1-2). Seventeen percent of the patients received tocilizumab, and 3% received tocilizumab and glucocorticoids. Neurologic events occurred in 6 patients within 4 weeks of infusion (grade 1, n = 4; grade 2, n = 2). The most common grade 1 or 2 neurologic events were tremor (18%), anxiety (11%), and disturbance in attention (11%). No CAR T-cell–related encephalopathy syndrome (CRES)37,38 of grade 3 or higher was confirmed in these 28 patients.
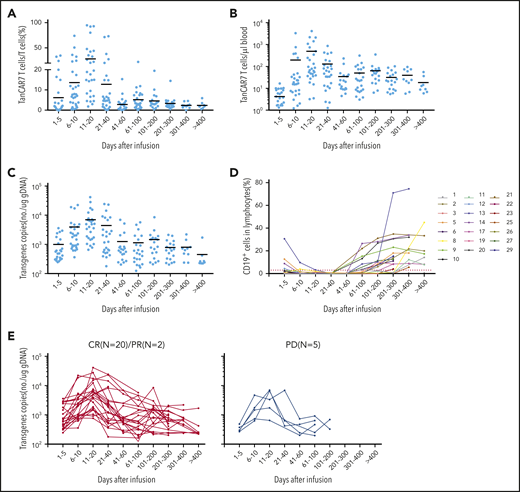
Peak expansion of TanCAR7 T cells occurred within the first 7 to 14 days postinfusion, and the mean number of circulating CAR T cells was 496 per microliter of blood (range, 3-4074). Low cell counts in peripheral blood were detectable by qPCR for up to 400 days in 8 patients with an ongoing CR (Figure 6A-C). Transient B-cell depletion occurred in all 22 patients who had a response, and 11 (50%) patients had sustained normal CD19+ B-cell recovery by ∼3 months after CAR T-cell infusion (Figure 6D). Serum cytokine levels were measured serially in all patients during the first month following therapy initiation. Among the cytokines detected, IL-2, IL-6, and IL-8 were elevated in the patients with CRS. Similar to previous reports on the effects of CD19 CAR T-cell therapy,18,37,39 the increase in IL-6 was significantly associated with CRS grade (supplemental Figure 12). Taken together, these data indicate that TanCAR7 T cells produce a potent and durable antitumor response.
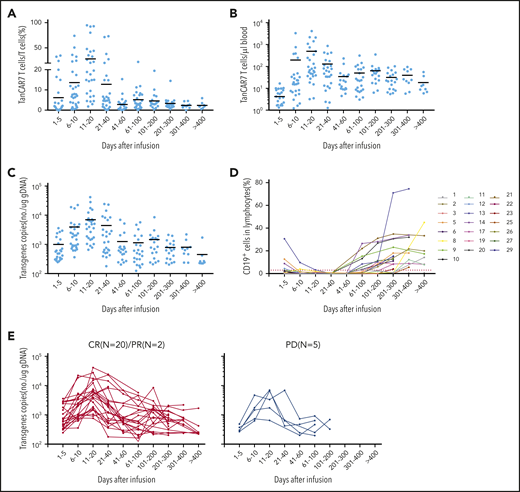
Expansion of peripheral blood CAR T cells and B-cell recovery. Percentage (A) and absolute number (B) of circulating TanCAR7 T cells, as determined by flow cytometry, and transgene copy number (C), as determined by qPCR. Horizontal solid lines show the mean at each time point. (D) Percentage of CD19+ cells in lymphocytes in blood. Red dotted line is 3%. (E) Individual concentration-time profiles in responding patients (left panel) and in nonresponding patients (right panel). gDNA, genomic DNA.
Expansion of peripheral blood CAR T cells and B-cell recovery. Percentage (A) and absolute number (B) of circulating TanCAR7 T cells, as determined by flow cytometry, and transgene copy number (C), as determined by qPCR. Horizontal solid lines show the mean at each time point. (D) Percentage of CD19+ cells in lymphocytes in blood. Red dotted line is 3%. (E) Individual concentration-time profiles in responding patients (left panel) and in nonresponding patients (right panel). gDNA, genomic DNA.
Discussion
Many studies have shown that CD19 is an effective immunotherapy target in r/rNHL as a result of its abundant, but restricted, expression in normal and malignant B cells.5-7 However, CD19− relapse has become a major challenge in the goal of achieving long-term disease remission after CD19 CAR T-cell therapy.8 Recently, dual targeting of CD19 and CD20 by CARs has been proposed to overcome CD19− relapse after CD19 CAR T-cell therapy.9,12,40 Similar to CD19, CD20 is expressed in normal B cells and associated malignancies but is not found on hematopoietic stem cells,41 which makes it 1 of the most promising treatment targets for B-cell malignancies. However, cotargeting multiple antigens expressed on malignant cells with diverse constructs, such as dual CAR and TanCAR constructs, remains in the preclinical testing stage. The infusion of dual-targeted CAR T cells may represent a feasible and reliable solution in clinical practice. Notably, the expression level of TanCARs is more uniform, and the aggregation of CAR molecules, the accumulation of F-actin, and the polarization of MTOC during IS formation were significantly better with TanCARs than with dual CARs.13 Therefore, in this study, we designed a series of TanCARs targeting CD19 and CD20 and found that TanCAR7 T cells showed dual antigen coverage and elicited a potent and durable antitumor response in vivo. Furthermore, we report the results of the first clinical trial of TanCAR targeting CD19 and CD20 T cells, which demonstrated its safety, potent clinical efficacy, low toxicity, and ability to potentially reduce antigen escape relapse in patients with r/rNHL.
The results of this study indicate that different scFv domains are critical for IS formation and signal transduction, which determine cell fate. We found that TanCAR7 T cells show dual antigen coverage of CD19 and CD20 and form superior and stable IS structures, which may be related to the stronger and more rapid degranulation observed in TanCAR7 T cells than in single-targeted CAR T cells. The cytolytic degranulation of perforin and granzyme is considered the main mechanism by which CAR T cells redirect the killing of target cells.23,42 In addition, cytokines secreted by CAR T cells play an important role in mediating tumor lysis through a secondary mechanism.43 Notably, although TanCAR7 forms a more stable IS structure, cytokine secretion after antigen stimulation was lower with TanCAR7 than with single-target CARs, especially CD20 CAR. We speculate that stable synaptic structures are merely triggering and necessary, but not sufficient, conditions for cytokine secretion. The regulation of cytokine secretion may involve the activation of secondary signaling pathways, such as those involving the transcription factors STAT3, NFAT, and NF-κB, after IS formation. Several results of this study indicate that TanCAR7 T cells retain markers associated with memory potential and express reduced levels of PD-1 and TIM-3, which may have contributed to the observed durable responses. Currently, our understanding of how TanCAR signaling directs T-cell functional outputs, such as robust antitumor activity and low toxicity, remains incomplete. Comprehensive mechanistic investigations of the signaling and activation features of CAR T cells are needed. In addition, all functional assays are made with bulk CAR T cells, rather than CD8+ and CD4+ subsets; further analyses will be performed to differentiate their contributions.
Among the enrolled patients, 82% had stage III or IV disease, 46% had an Eastern Cooperative Oncology Group performance status score of 2, 25% had bulky disease, 64% were positive for Ki-67 in >70% of lesions at baseline, 61% had extranodal organ involvement, and 86% had refractory disease. Although many patients in our study were diagnosed with relapsed or refractory disease, an extremely high tumor burden, and adverse cytogenetic or genomic aberrations, impressive responses were achieved. The overall response rate was 79%, and the CR rate was 71%. Progression-free survival was 64% at 12 months. In particular, antigen loss relapse was observed in only 1 patient, whereas CD19 and CD20 double-positive relapse occurred in the remaining patients after TanCAR7 T-cell infusion. Importantly, no case of defined CRES of grade 3 or higher was confirmed in any patient. Nearly all of the high-grade CRS and CRES cases were reversible, and the incidence rate was lower than that reported previously. Thus, TanCAR7 T-cell therapy is a safe and efficient approach to overcome CD19 or CD20 antigen loss.
Currently, there are different views on whether long-term clinical efficacy requires the persistence of CAR T cells in patients with NHL. In our clinical trial, we found that CAR T cells were detectable for a longer duration in patients who achieved a clinical response than in those without a response (Figure 6E). In general, if CAR T-cell therapy is used as a definitive therapy rather than a bridging therapy, there should be considerable effort to extend the half-life of CAR T cells in vivo to ensure durable efficacy, in addition to avoiding recurrence due to antigen loss. In the present study, the peak expansion of TanCAR7 T cells occurred within the first 7 to 14 days postinfusion, the mean number of circulating CAR T cells was 496 per microliter of blood, and low levels of these cells were detectable by qPCR analysis for up to 400 days in 8 patients with an ongoing CR. These clinical data show that TanCAR7 T cells exert a durable antitumor response with a high expansion peak in r/rNHL.
In summary, this study shows that TanCAR7 T cells evoke a potent and durable antitumor response, and no case of grade 3 or higher CRES was observed during the treatment of r/rNHL.
The RNA sequencing data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE134937).
Data sharing requests should be sent to Weidong Han (hanwdrsw69@yahoo.com).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Yuche Biotechnology core facilities, including the bioinformatics core and the integrated genomics operation core, for excellent support.
This work was supported, in part, by the National Natural Science Foundation of China (31870873, 81830002, 81602711, and 31991171), the Leading Talents Grant of Science & Technology from Beijing (Z181100006318004), the National Key Research and Development Program of China (2016YFC1303501 and 2016YFC1303504 [W.H.], 2017YFC0909803 [W.Y.]), and a grant from the Key Nursery Project of Chinese PLA General Hospital (16KMZ05).
Authorship
Contribution: C.T., Y.W., Z.W., and W.H. designed the research; C.T., Y.W., X.J., H.D., and Y.G. performed experiments; X.H. and D.T. assisted with experiments; C.T., Y.Z., Y.L., W.Z., C.W., Q.Y., and W.H. performed the clinical trial; and C.T., Y.W., Y.Z., W.Z., Z.W., W.L., and W.H. analyzed the data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Weidong Han, Chinese PLA General Hospital, No. 28 Fuxing Rd, Haidian District, Beijing 100853, China; e-mail: hanwdrsw69@yahoo.com.