


Abstract
Acquired genetic mutations in hematopoietic stem or progenitor cells can lead to clonal expansion and imbalanced blood cell production. Clonal hematopoiesis is exceptionally common with human aging, confers a risk of evolution to overt hematologic malignancy, and increases all-cause mortality and the risk of cardiovascular disease. The degree of risk depends on the specific mutant allele driving clonal expansion, number of mutations, mutant allele burden, and concomitant nongenetic risk factors (eg, hypertension or cigarette smoking). People with clonal hematopoiesis may come to clinical attention in a variety of ways, including during the evaluation of a possible hematologic malignancy, as an incidental discovery during molecular analysis of a nonhematologic neoplasm, after hematopoietic cell transplantation, or as a result of germline testing for inherited variants. Even though the risk of clonal progression or a cardiovascular event in an individual patient with clonal hematopoiesis may be low, the possibility of future clinical consequences may contribute to uncertainty and worry, because it is not yet known how to modify these risks. This review summarizes clinical considerations for patients with clonal hematopoiesis, including important points for hematologists to consider discussing with affected persons who may understandably be anxious about having a mutation in their blood that predisposes them to develop a malignancy, but which is significantly more likely to result in a myocardial infarction or stroke. The increasing frequency with which people with clonal hematopoiesis are discovered and the need for counseling these patients is driving many institutions to create specialized clinics. We describe our own experience with forming such clinics.
Introduction
Clonal hematopoiesis, defined as an outsized contribution to circulating blood cell production by a single genetically altered hematopoietic clone in the absence of diagnostic evidence of a hematologic neoplasm, is a common biological state in middle-aged and older persons.1-4 Clonal hematopoiesis usually results from acquisition by a hematopoietic stem or progenitor cell of one or more of a limited repertoire of somatic mutations, although somatic mosaicism and imbalance in hematopoiesis can also result from large structural chromosomal rearrangements.5,6 Aging-associated clonal hematopoiesis is a risk factor for further mutation acquisition and clonal evolution to an overt hematologic neoplasm (Table 1), as well as a risk factor for all-cause mortality and specifically death from a cardiovascular event (eg, myocardial infarction or cerebrovascular accident).1,7
Hematologic malignancies observed in representative studies of clonal hematopoiesis
| Reference | Sequencing approach | Development of hematologic cancer with clonal hematopoiesis, HR (95% CI) | Cases of hematologic malignancy, n | Malignancy types | Hematologic malignancies in patients with preexisting clonal hematopoiesis, % |
|---|---|---|---|---|---|
| 2 | Whole exome | 11.1 (3.9-32.6) | 16 | 6 lymphoma NOS; 4 leukemia NOS; 2 unspecified; 2 MM; 1 MDS;1 AML | 31 |
| 3 | Whole exome | 12.9 (5.8-28.7) | 37 | 22 not listed; 3 CLL; 2 MDS; 2 MPN NOS; 2 AML; 1 lymphoma NOS; 2 MM or other plasma cell neoplasm; 1 CMML; 1 acute leukemia NOS; 1 chronic leukemia NOS | 42 |
| 5 | SNP array | 35.4 (14.7-76.6) | 43 | Incident hematologic cancer diagnoses not specified | NA |
| 6 | SNP array | 10.1 (5.8-17.7) | 105 | 38 lymphoma; 19 MM; 14 MDS; 10 CLL; 7 AML; 4 lymphoid leukemia NOS; 3 MPN NOS 3 myeloid leukemia NOS; 2 CMML; 1 hairy cell leukemia; 1 MF; 1 CML; 1 ALL; 1 leukemia NOS | 14 |
| Reference | Sequencing approach | Development of hematologic cancer with clonal hematopoiesis, HR (95% CI) | Cases of hematologic malignancy, n | Malignancy types | Hematologic malignancies in patients with preexisting clonal hematopoiesis, % |
|---|---|---|---|---|---|
| 2 | Whole exome | 11.1 (3.9-32.6) | 16 | 6 lymphoma NOS; 4 leukemia NOS; 2 unspecified; 2 MM; 1 MDS;1 AML | 31 |
| 3 | Whole exome | 12.9 (5.8-28.7) | 37 | 22 not listed; 3 CLL; 2 MDS; 2 MPN NOS; 2 AML; 1 lymphoma NOS; 2 MM or other plasma cell neoplasm; 1 CMML; 1 acute leukemia NOS; 1 chronic leukemia NOS | 42 |
| 5 | SNP array | 35.4 (14.7-76.6) | 43 | Incident hematologic cancer diagnoses not specified | NA |
| 6 | SNP array | 10.1 (5.8-17.7) | 105 | 38 lymphoma; 19 MM; 14 MDS; 10 CLL; 7 AML; 4 lymphoid leukemia NOS; 3 MPN NOS 3 myeloid leukemia NOS; 2 CMML; 1 hairy cell leukemia; 1 MF; 1 CML; 1 ALL; 1 leukemia NOS | 14 |
ALL, acute lymphoid leukemia; CLL, chronic lymphoid leukemia CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; MF, myelofibrosis; MM, multiple myeloma; MPN, myeloproliferative neoplasm; NOS, not otherwise specified; SNP, single-nucleotide polymorphism.
When a somatic mutation leading to clonal expansion occurs in a leukemia-associated gene, and the variant allele frequency (VAF; ie, proportion of mutant DNA) of that mutation is at least 2% (ie, more than 4% of circulating blood cells are derived from a single clone, if heterozygosity and a diploid state are assumed), the term “clonal hematopoiesis of indeterminant potential” (CHIP) can be used.8,9 CHIP indicates that the consequence of clonal hematopoiesis for the individual is unknown: such clones usually have no clinical consequence, but they have the potential to further expand or evolve into overt neoplasia. Alternatively, a clone can contribute to a vascular event by a proinflammatory and proatherogenic interaction with endothelium,7,10-12 can worsen heart failure by altering myocardial remodeling,13 or can potentiate other nonneoplastic pathology.14,15
CHIP is present in at least 10% to 20% of people by age 70 years.2,3 Smaller expanded hematopoietic clones that do not meet the proposed definition of CHIP can be detected with highly sensitive, error-corrected sequencing methods in almost every individual by age 50, but the clinical consequences of smaller clones are less clear.16,17 Age-related clonal hematopoiesis (ARCH) is a term used by some clinicians and investigators to emphasize that emergence of somatically mutated hematopoietic clones is an almost universal part of the aging process, and that most clones do not progress to hematologic malignancy. CHIP, in contrast to ARCH, requires a specific mutant allele burden and that a somatic mutation emerge in a gene associated with hematologic neoplasia. ARCH can be said to include CHIP, as well as other forms of clonal hematopoiesis that are probably of less clinical significance.
Increasingly, for a variety of reasons, CHIP is detected before a hematologic malignancy or cardiovascular event occurs. How to counsel people with clonal hematopoiesis or monitor them prospectively is currently an area of uncertainty. The authors each lead new specialized clinics for counseling patients with CHIP and other precursor conditions that have a risk of evolution to hematologic neoplasia (Figure 1). We summarize herein some key biological and clinical observations with respect to clonal hematopoiesis based on published literature and our personal experiences.
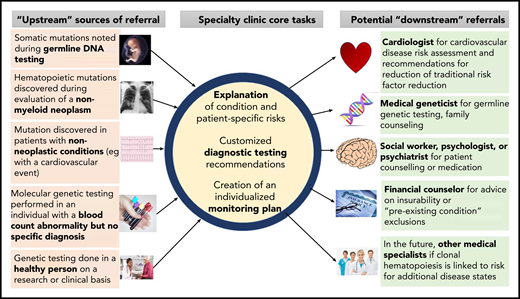
Referral patterns and evaluation of people with clonal hematopoiesis. People with clonal hematopoiesis may be identified in several different ways, including during testing for other conditions. Tasks for hematologists include helping affected persons understand clonal hematopoiesis and its implications, organizing further diagnostic testing that may be indicated in some cases (eg, bone marrow aspiration and biopsy), and developing a monitoring plan. Referral to other specialists may be necessary, including geneticists (eg, if a germline mutation is possible) or cardiovascular disease specialists for traditional risk factor assessment and modification.
Referral patterns and evaluation of people with clonal hematopoiesis. People with clonal hematopoiesis may be identified in several different ways, including during testing for other conditions. Tasks for hematologists include helping affected persons understand clonal hematopoiesis and its implications, organizing further diagnostic testing that may be indicated in some cases (eg, bone marrow aspiration and biopsy), and developing a monitoring plan. Referral to other specialists may be necessary, including geneticists (eg, if a germline mutation is possible) or cardiovascular disease specialists for traditional risk factor assessment and modification.
The following 5 scenarios indicate some of the ways in which patients with clonal hematopoiesis may be referred to a hematologist or to a specialty clinic.
Case 1
A 49-year-old premenopausal woman was diagnosed with a node-negative, hormone-receptor–negative, 1.5-cm ductal carcinoma of the right breast. Local resection of the tumor, adjuvant chemotherapy, and radiotherapy were recommended by her oncologist. Because her mother had a history of invasive breast cancer and a maternal aunt a history of ductal carcinoma in situ, germline genetic testing was recommended and was performed on blood-derived DNA, using a commercially available targeted sequencing panel focused on inherited variants predisposing to breast cancer, including BRCA1, BRCA2, CHEK2, ATM, and TP53. The genetic testing laboratory reported that germline variants were not detected in the patient, but a TP53 p.Y234C mutation was observed with a VAF of 9%. Because most heterozygous germline gene polymorphisms or mutations are present at a VAF of 40% to 60% in the absence of loss of heterozygosity, the testing laboratory reported that the TP53 variant was most likely to be somatic and acquired, rather than germline.
Case 2
During evaluation of newly diagnosed Waldenström macroglobulinemia (WM), a 61-year-old man underwent bone marrow aspiration and biopsy. The patient had been healthy except for hypertension. Testing of his marrow mononuclear cells included next-generation sequencing with a targeted panel. The institution at which he was assessed used a single 95-gene panel assay for all hematologic malignancies, which simplified electronic test ordering and laboratory workflow. In addition to an MYD88 mutation18 characteristic of WM with a VAF of 8%, a TET2 frameshift mutation was noted with a VAF of 21%. His marrow showed no evidence of myelodysplastic syndromes (MDSs) or other myeloid neoplasias and 15% involvement by lymphoplasmacytic lymphoma.
Case 3
A 71-year-old woman was diagnosed with squamous cell lung cancer with unilateral hilar adenopathy on radiography. As part of her staging evaluation, she underwent testing with a developmental cell-free/circulating tumor DNA assay that included genes that were not restricted to those commonly associated with lung cancer. The patient was incidentally noted to have JAK2 p.V617F mutation with a VAF of 3%. The patient’s blood counts were normal, her spleen was not enlarged, and she had no history of thrombosis or constitutional symptoms.
Case 4
A 73-year-old man who is an emeritus professor of oncology at a major medical school read several papers about clonal hematopoiesis and asked his primary care physician to test him for CHIP. The patient had a normal complete blood count other than a slightly abnormal red cell distribution width (15.3%; laboratory normal range 11% to 14.5%). The primary care physician deferred to the oncologist’s perceived broader knowledge base and ordered a gene-sequencing panel for common CHIP-associated genes. A DNMT3A p.R882H mutation was detected at a VAF of 7%.
Case 5
A 68-year-old woman with high-risk MDS received an allogeneic hematopoietic cell transplant from her 66-year-old fully matched brother, who had a normal blood counts before the cells were harvested. After the transplantation, full male donor chimerism was achieved, but the patient had persistent cytopenias that remained unexplained despite extensive evaluation. Marrow biopsy showed mild hypercellularity for age without dysplasia, and molecular genetic testing showed an ASXL1 nonsense mutation with a VAF of 16% that was not present in the patient’s blood before the transplantation. During subsequent evaluation, the ASXL1 mutation was found in a blood sample from the donor as well, with a lower VAF (3%).
How does clonal hematopoiesis arise?
Somatic DNA mutations accumulate in every tissue of the body during aging.19-22 In hematopoietic stem cells, exonic mutations occur on the order of 1 mutation per decade of life, and a small subset of these provide a fitness advantage and result in clonal expansion.23 Because blood cells circulate in large numbers, whereas cells derived from other tissues subject to greater anatomical constraints do not, clonal hematopoiesis has distinct clinical implications compared with nonhematologic somatic mosaicism.1,24
The most common biochemical mutational event giving rise to clonal hematopoiesis is spontaneous deamination of methylated cytosine at CpG dinucleotides resulting in generation of thymine, which is not appropriately repaired and is then stably passed on to daughter cells.25 Stable DNA alterations related to nonhomologous end joining and large chromosomal structural rearrangements also occur. Murine models have been helpful in illuminating the precise mechanisms by which some of these mutations result in clonal expansion, but they incompletely model clonal hematopoiesis, especially that associated with splicing mutations.26
The population prevalence of clonal hematopoiesis depends on the detection technique used. Currently used whole-exome and whole-genome approaches are insensitive for detecting clones with VAFs <5%, whereas targeted sequencing panels can routinely detect mutations down to VAFs of 1% to 2%. Using high-sensitivity, error-corrected targeted sequencing methods, clones with VAFs of <0.1% can be found, and virtually all individuals will have evidence of this degree of clonal expansion by age 50 years.1 However, the clinical significance of very small (<1% VAF) clones is unclear. Clones with >10% VAFs and that are associated with a splicing mutation or with more than one leukemia-associated driver mutation are associated with an increased risk of clonal progression, compared with smaller clones or those defined by a single mutation.27
Who is at risk for clonal hematopoiesis?
Because mutation acquisition is cumulative and time dependent, the dominant risk factor for clonal hematopoiesis is aging, as it is for most myeloid and many lymphoid neoplasms.28 Modest increases in the prevalence of CHIP have been described in males, individuals of Hispanic ethnicity, and smokers.2,3 Some somatic mutations are more likely to be observed in specific clinical settings (Table 2).
Clonally restricted mutations observed in specific clinical settings in the absence of overt neoplasia
| Clinical setting and reference(s) | Gene |
|---|---|
| Aging-associated: ARCH or CHIP2-4,16,17 | DNMT3A, TET2, ASXL1, JAK2, TP53; many others are recurrent but less frequent |
| History of chemotherapy or radiotherapy35,36 | TP53, PPM1D |
| Aplastic anemia34 | BCOR, BCORL1, PIGA; DNMT3A, ASXL1 |
| Severe congenital neutropenia or Shwachman-Bodian-Diamond syndrome73,74 | CSF3R, TP53 |
| Unexplained monocytosis75 | ASXL1, CBL, DNMT3A, NRAS, RUNX1 |
| Clinical setting and reference(s) | Gene |
|---|---|
| Aging-associated: ARCH or CHIP2-4,16,17 | DNMT3A, TET2, ASXL1, JAK2, TP53; many others are recurrent but less frequent |
| History of chemotherapy or radiotherapy35,36 | TP53, PPM1D |
| Aplastic anemia34 | BCOR, BCORL1, PIGA; DNMT3A, ASXL1 |
| Severe congenital neutropenia or Shwachman-Bodian-Diamond syndrome73,74 | CSF3R, TP53 |
| Unexplained monocytosis75 | ASXL1, CBL, DNMT3A, NRAS, RUNX1 |
Germline loss of the MBD4 gene encoding the enzyme methyl-CpG binding domain 4 DNA glycosylase, important for repair of cytosine to thymine DNA transitions, leads both to an increased likelihood of clonal hematopoiesis and MDS or acute myeloid leukemia (AML) and to a markedly increased C to T mutation burden when AML develops.29 There are likely to be numerous germline predispositions to clonal hematopoiesis other than MBD4 loss, which is rare. An intronic polymorphism in the TERT gene encoding telomerase reverse transcriptase, for example, is associated with an increased risk of clonal hematopoiesis, including clonal hematopoiesis measured by a somatic mutation burden outlier state detected by whole-genome sequencing rather than a leukemia driver mutation state.30 Polymorphisms in MPL, FRA10B, and TM2D3-TARSL2 are associated with somatic mosaicism at the autosomal chromosomal level.31
Recently, 156 germline genetic determinants of acquired loss of chromosome Y in males were identified in the UK Biobank population (∼205 000 persons) and then validated in 757 114 men of Japanese or European ancestry. These variants were enriched for genes encoding factors involved in cell cycle regulation and cancer susceptibility.32 Furthermore, in a study of 500 sibling allogeneic hematopoietic cell transplant donors aged ≥55 years, 16% of donors had CHIP, with a median VAF of 5.9%: 19.2% of donors for recipients with myeloid neoplasm, compared with only 6.3% of donors for siblings with lymphoid malignancies.33 Given the relative myeloid bias in malignancies arising from CHIP in some series, this suggests a common predisposition (genetic or environmental) to clonal hematopoiesis in sibling pairs.
Individuals with immune-mediated marrow failure frequently have clonal hematopoiesis, including mutations in PIGA, which is associated with paroxysmal nocturnal hemoglobinuria.34 A special case is clonal hematopoiesis with somatic variants in TP53 or PPM1D, as such preexisting clones are strongly selected for in patients undergoing cytotoxic chemotherapy or radiotherapy.35-37 Clonal hematopoiesis marked by these genes at the time of cytotoxic therapy is a major risk factor for subsequent development of therapy-related MDS (t-MDS)/AML.38,39 In the future, knowledge of the presence of preexisting TP53 mutant clones may influence decision-making about adjuvant therapy.40
What are the clinical consequences of clonal hematopoiesis?
It is important to keep in mind that many people with clonal hematopoiesis will experience no clinical consequences; thus, CHIP could be considered a biological state that can be a risk factor for disease, not a disease in itself. Acquisition of a secondary driver gene mutation may result in progression to overt malignancy.41 Although the relative risk for myeloid disease in patients meeting the definition of CHIP is high (>10-fold), in part because of the low incidence of these neoplasms in the general population, the absolute risk has been estimated at between 0.5% and 1% per year.2,3
Clonal hematopoiesis is associated with poorer outcomes after autologous transplantation in some settings.42 In the allogeneic transplantation setting, use of a donor with CHIP was associated with unexplained cytopenias in 1 series43 and with more frequent development of chronic graft-versus-host disease (hazard ratio [HR], 1.7; 95% confidence interval [CI], 1.2-2.5) and higher nonrelapse mortality, but lower malignancy relapse rate (HR, 0.6; 95% CI, 0.4-0.9), such that there was not a clear effect of donor CHIP status on survival in another series.33 It is possible that donors with clonal hematopoiesis are a risk factor for donor-derived leukemias, but it is difficult to eliminate a contribution from an abnormal recipient microenvironment that permits clonal outgrowth.44,45
Patients with aplastic anemia harboring certain types of clones (ie, with mutations other than PIGA, BCOR, or BCORL1) have a poorer prognosis, higher rate of evolution to MDS or AML, and lower response to anti-T-cell–immunosuppressive therapy.34 JAK2 is the fourth or fifth most commonly mutated gene in clonal hematopoiesis, as in “Case 3,” and is associated with an increased risk of arterial and venous thrombosis in addition to evolution to an overt myeloproliferative neoplasm.46 JAK2 and other blood compartment–restricted mutations (eg, KRAS) are recurrently detected in patients with nonhematologic neoplasms with commercially available cell-free/circulating tumor DNA assays.47
Patients with unexplained cytopenias despite thorough hematology evaluation including marrow examination are frequently said to have idiopathic cytopenias of undetermined significance (ICUS).48-50 Although assessment of unexplained cytopenias is beyond the scope of this review, patients with unexplained cytopenias in association with clonal hematopoiesis (clonal cytopenias of undetermined significance [CCUS]) have a markedly increased risk of progression to MDS or AML diagnosed according to World Health Organization criteria, compared with those with ICUS without a clonal marker.51 For example, Malcovati and colleagues reported results obtained from long-term follow-up of patients with cytopenias who had a nondiagnostic bone marrow aspirate and biopsy at initial evaluation. The investigators showed that the presence of a clonal mutation was highly predictive of the risk of transformation to hematologic malignancy (HR, 13.9; 95% CI, 5.4-35.9; 5- and 10-year cumulative probabilities of progression, 82% for CCUS vs 9% for ICUS and 95% vs 9%, respectively).51
Strikingly, patients with clonal hematopoiesis have an increased risk of cardiovascular events (HR, 1.9; 95% CI, 1.4-2.7), and clonal hematopoiesis carries a similar order of magnitude of cardiac risk as traditional risk factors, such as smoking, hyperlipidemia, and hypertension.7,12,52 The mechanism by which clonal hematopoiesis contributes to myocardial infarction and stroke is thought to be proinflammatory, proatherogenic interactions between circulating clonal monocytes/macrophages and the endothelium or by nascent atherogenic plaques. This process can be blocked in preclinical models by inhibitors of the NLRP3 inflammasome.7,10 Clonal hematopoiesis is also associated with worse clinical outcomes in the setting of congestive heart failure,13 probably because of altered ventricular remodeling by infiltrated clonal monocytes/macrophages, also in an NLRP3-dependent fashion. Other clinical associations between nonhematologic disease and clonal hematopoiesis are being sought by multiple research groups.53 Murine models suggest that the gut microbiome influences the risk of clonal progression.54
Who should be notified of clonal hematopoiesis?
In the absence of established interventions to eliminate expanded clones, the benefit of testing for and informing patients of an incidental finding of clonal hematopoiesis is still unclear, particularly when considering the potential psychological impact of such an unmodifiable risk factor for disease. Currently, we cannot recommend universal notification of patients about all hematopoietic clones, given that many clones will be of no consequence.
However, there are some settings where notification should be considered. For example, individuals may be found to have CHIP with clinical or mutational features associated with higher risk of hematologic malignancy, such as abnormal blood count indices or high-risk mutational characteristics (chromosomal aneuploidy, higher VAF of somatic mutations, or more than 1 known myeloid neoplasm driver mutation, especially in higher risk genes such as IDH1/2, TP53, or spliceosome components). In these settings, we recommend that patients and their care team consider notification, especially if evaluation for an occult hematologic disorder might be warranted. In addition, given the high risk of cardiovascular disease or thrombosis conferred by the JAK2 V617F mutation, disclosure of CHIP related to this mutation should be strongly considered. The decision to notify individuals about CHIP should take into account the patient’s life expectancy, personal preferences, and local cultural context. We recommend that the potential to discover clonal hematopoiesis be included in consent discussions for genetic testing whenever possible and that individuals be given the option to be or not be informed of CHIP as an incidental finding.
How might clinical consequences of clonal hematopoiesis be averted?
Elimination of expanded clones by a targeted therapy or selective immunotherapy to prevent subsequent evolution to a neoplasm is not yet feasible, but is an attractive goal. However, given the relatively low rate of neoplastic progression or other clinical consequences of clonal hematopoiesis, adverse effects of treatment aimed at clone elimination must be carefully considered and may prove to be justified in only certain cases.
Existing therapies for myeloid neoplasia, such as DNA hypomethylating agents or lenalidomide, are unlikely to be selective enough or have a favorable risk-benefit balance when used in the setting of most CHIP cases, but these drugs may eventually be found to reduce overall clonal burden and delay disease onset in certain cases with large clones, and that could ultimately be beneficial. Among targeted agents, splicing inhibitors (eg, E782055 and H3B-880056 ) or IDH inhibitors are attractive, although splicing and IDH mutations are far less common CHIP-associated variants than DNMT3A, TET2, or ASXL1. An interventional trial of intravenous vitamin C in TET2 mutant CCUS (NCT 03682029) is ongoing, prompted by the observation that TET2 function can be restored and aberrant leukemic stem cell self-renewal can be disrupted with high concentrations of vitamin C in preclinical models. Orally administered vitamin C, in contrast, does not typically achieve a high enough concentration to alter TET2 function meaningfully.57
From a public health standpoint, the cardiovascular risk associated with clonal hematopoiesis is of greater consequence than relatively rare neoplastic progression.52 Anti-inflammatory approaches may be helpful in preventing cardiac events. Atherosclerosis has long been recognized as an inflammatory disease,58,59 and clonal hematopoiesis may provide a mechanism that links inflammation and atherosclerosis. In a randomized placebo-controlled trial (CANTOS) of the anti-interleukin 1β antibody canakinumab in 10 061 patients who had a history of myocardial infarction and had elevated C-reactive protein, canakinumab prevented recurrent cardiovascular events and stroke.60 A post hoc sequencing analysis of pretreatment samples from nearly 4000 patients enrolled in CANTOS found that this benefit was largely confined to subjects with CHIP, especially TET2-mutant CHIP, which was (perhaps not coincidentally, as DNMT3A may drive inflammation to a lesser degree61 ) the most common clonal mutation in this postmyocardial infarction population.62 More recently, a placebo-controlled study of the antimacrophage agent colchicine in 4745 patients with a history of myocardial infarction also showed benefit in preventing recurrent cardiac events. These colchicine-treated patients have not yet been analyzed for clonal hematopoiesis.63
For now, monitoring of blood counts and control of recognized risk factors for cardiac disease are the main approach to patients with clonal hematopoiesis. Key questions remain unresolved. For example, which patients should undergo marrow aspiration at the time of initial assessment, and what is the optimal frequency of prospective blood count monitoring depending on the patient’s specific progression risk?40 It seems that a patient with CCUS and multiple high-VAF mutations that include a splicing variant should be monitored with more frequency than, for instance, someone with only a DNMT3A non-R882 mutant clone of 2.5% VAF, but there is no consensus on specific approaches.
In addition, the optimal lipid and blood pressure goals for patients with clonal hematopoiesis, and which patients should undergo additional exercise stress testing or computed tomography coronary calcification assessment, remain uncertainties. Increasingly, patients with clonal hematopoiesis will be candidates for interventional clinical trials to mitigate both hematologic and cardiovascular risk.
Finally, as with other conditions in which a “watchful waiting” or active surveillance approach is undertaken,64 a subset of patients will understandably become anxious or worried when learning about clonal hematopoiesis. Patients may also worry about losing eligibility for life or health insurance, or about the logistics of monitoring. Having a plan for psychosocial and other assistance of those with higher levels of anxiety is essential.
How to create a CHIP clinic
Some institutions are now considering creating specialty clinics for assessment of patients with clonal hematopoiesis, and the clinicians and administrators undertaking this effort are encountering recurrent challenges. We can learn from the experiences of others.
Because there may be uncertainty in some cases about whether a detected variant is germline or somatic,65,66 especially with high VAF TP53 mutations (>40%), access to geneticists who can arrange for testing of nonhematopoietic tissue (eg, by skin biopsy and creation of a fibroblast cell line as a germline control) and address nonhematologic consequences of germline variants and familial considerations is important. Likewise, collaboration with cardiovascular specialists is essential, given the high risk of cardiovascular events in patients with CHIP. Growth in the field of cardio-oncology may facilitate referral. If other disease states, such as autoimmune conditions or neurodegenerative disorders, turn out to be increased in persons with CHIP, then close collaboration with specialists in other groups may become necessary, as well.
In some institutional and clinical settings, especially where genetic testing is not commonly performed, there may not be enough patients yet to justify creation of a specific clinic or service dedicated to CHIP and related states. In these settings, partnerships with hematologists interested in other malignancy precursor states (eg, monoclonal B-cell lymphocytosis or monoclonal gammopathy of undetermined significance [MGUS]) may help secure adequate institutional resources and assure a more stable and predictable referral population.
At Dana-Farber Cancer Institute (DFCI), for example, we have partnered with colleagues who are formally studying monoclonal gammopathies and the transition from MGUS to smoldering myeloma to multiple myeloma,67 a process that has some parallels with CHIP as a precursor state to malignancy that can cause nononcologic problems (eg, amyloidosis, metabolic bone disease, or renal injury in the case of MGUS/smoldering myeloma), to create a Center for Prevention of Progression (CPOP) of Hematologic Malignancies, locally called the Precursor Clinic.68 We have benefitted from collaboration of an enthusiastic group of cardiologists at Brigham & Women’s Hospital who have a long-standing interest in atherosclerosis as an inflammatory disease,58 as well as a large and experienced cancer genetics clinical group.
At Memorial Sloan Kettering Cancer Center (MSKCC), we routinely perform parallel sequencing, in which a primary tumor (eg, solid tumor) is sequenced while blood is sequenced as a control to rule out germline variants including rare single-nucleotide polymorphisms. (This approach is not standard at DFCI.) Because up to 30% of individuals tested have clonal hematopoiesis, a substantial number of patients are referred by solid tumor specialists for hematology assessment.36,69 In addition, patients with nonhematologic neoplasms are commonly seen by our hematology or leukemia services for evaluation of prolonged or pronounced cytopenias in the setting of oncologic therapy. Assessment for acquired mutations associated with myeloid neoplasia is frequently performed and commonly reveals clonal hematopoiesis. As at DFCI, at MSKCC we focus on management of cardiovascular risk factors as well as blood count monitoring and have developed an algorithm for management of CHIP in patients with solid tumors.40 We are developing genotype-specific trials for subtypes of clonal hematopoiesis.
Billing and coding considerations
From a practical standpoint, there is no International Statistical Classification of Diseases and Related Health Problems (ICD) code for clonal hematopoiesis (at least in the version currently most widely used in the United States, ICD-10-CM), and this absence of a billable code may influence reimbursement for consultation of patients with CHIP. If evaluated patients have cytopenia, they can be classified accordingly, but those in whom CHIP has been identified may not fall into a specific category. We sometimes code patients using “Z15.09: Genetic susceptibility to other neoplasm” or “Z15.89: Genetic susceptibility to other disease,” and, in our experience, reimbursement rates have been high with this approach. However, a genetic susceptibility code is not usually enough to justify cardiology referral. The economics and logistics of DNA sequencing in clinical practice are beyond the scope of this review, but deserve careful consideration as well.
Management of described cases
Case 1
This 49-year-old woman with breast cancer reported that she was told by her treating medical oncologist that, because the detected TP53 variant was not germline, it was of no clinical significance. Therefore, the patient underwent planned adjuvant chemotherapy and experienced more cytopenias than expected, although hemoglobin and neutrophil counts eventually recovered to normal. She then self-referred herself for hematology consultation after completion of adjuvant radiotherapy, at which time the VAF of the TP53 mutation had increased to >30% and she had an elevated mean corpuscular volume and persistent mild thrombocytopenia. This patient’s expected increment of survival from the adjuvant therapy for breast cancer is <5%, whereas the likelihood that development of t-MDS/AML was accelerated by the adjuvant therapy is greater than that.4,36,38 In the future, we anticipate that information about clonal hematopoiesis could be part of informed discussion with patients about the risks and benefits of adjuvant therapy.
Case 2
In the patient with newly diagnosed WM and a TET2 mutant clone that is likely to be distinct from the WM clone, with normal blood counts and cell morphology, the patient has been monitored with blood counts 1 to 2 times per year. Blood counts have remained normal after 3 years. His hypertension has been optimally controlled. He underwent an elective cardiac exercise stress test without any evidence of ischemia.
Case 3
This patient, who had lung cancer and an incidentally discovered JAK2 mutation, underwent successful surgical excision of her primary tumor with careful attention to venous thromboembolism prophylaxis perioperatively. The serum erythropoietin level was normal. She elected to take low-dose aspirin indefinitely but was not thought to have an indication for cytoreductive therapy. Her blood counts are being monitored periodically, and she has not experienced a complication at this writing.
Case 4
The emeritus professor with a DNMT3A mutation has struggled with fears about his future. He has advised several of his colleagues not to undergo genetic testing, saying, “Sometimes it is better not to know.”
Case 5
The patient with donor-derived clonal hematopoiesis after receiving an allogeneic transplant from an older sibling has been managed expectantly. If there is clonal progression and a second transplantation has to be undertaken, an alternative donor will be sought. Transplantation programs differ in their approach to screening older donors for CHIP; this complex topic is the subject of Point-Counterpoint articles in Blood Advances.70,71 Although CHIP is more common in older donors, recent data indicate that small clones can be detected with sensitive error-corrected sequencing techniques in a large proportion of younger donors (clones with a median VAF of 0.00247 were found in 44% of 25 donors with a median age of 36 years), and these clones usually engraft in the recipient and expand over time, yet donor-derived leukemia is rare.72
Conclusion
Clonal hematopoiesis is increasingly recognized and carries a risk of both clonal progression and cardiovascular morbidity and mortality. Factors contributing to initiation of clonal expansion and drivers of clonal evolution are incompletely understood. The optimal management of affected persons, beyond blood count monitoring and control of cardiovascular risk factors, remains unclear and is an area of active investigation.
Acknowledgments
D.P.S. is supported by the Edward P. Evans Foundation; National Institutes of Health, National Cancer Institute (NCI) SPORE grant P50 CA206963 (principal investigators: Benjamin L. Ebert and Richard M. Stone); the James and Lois Champy Fund; and the Russell Hartmann Memorial Fund. K.L.B. is supported by the Edward P. Evans Foundation; NIH, NCI grant K08 CA241318-01; and the American Society of Hematology.
Authorship
Contribution: D.P.S. and K.L.B. wrote the article.
Conflict-of-interest disclosure: D.P.S. has served on data safety monitoring committees and consulting related to clinical trials for Celgene, Daiichi Sankyo, Janssen, Onconova, and Otsuka and his institution has received funds to support the conduct of clinical trials from Aprea and H3 Biosciences. K.L.B. declares no competing financial interests.
Correspondence: David P. Steensma, Dana-Farber Cancer Institute, D2037, 450 Brookline Ave, Boston, MA 02215; e-mail: david_steensma@dfci.harvard.edu.
