


Abstract
Inherited bone marrow failure syndromes (IBMFSs) are characterized by ineffective hematopoiesis and increased risk for developing myeloid malignancy. The pathophysiologies of different IBMFSs are variable and can relate to defects in diverse biological processes, including DNA damage repair (Fanconi anemia), telomere maintenance (dyskeratosis congenita), and ribosome biogenesis (Diamond-Blackfan anemia, Shwachman-Diamond syndrome). Somatic mutations leading to clonal hematopoiesis have been described in IBMFSs, but the distinct mechanisms by which mutations drive clonal advantage in each disease and their associations with leukemia risk are not well understood. Clinical observations and laboratory models of IBMFSs suggest that the germline deficiencies establish a qualitatively impaired functional state at baseline. In this context, somatic alterations can promote clonal hematopoiesis by improving the competitive fitness of specific hematopoietic stem cell clones. Some somatic alterations relieve baseline fitness constraints by normalizing the underlying germline deficit through direct reversion or indirect compensation, whereas others do so by subverting senescence or tumor-suppressor pathways. Clones with normalizing somatic mutations may have limited transformation potential that is due to retention of functionally intact fitness-sensing and tumor-suppressor pathways, whereas those with mutations that impair cellular elimination may have increased risk for malignant transformation that is due to subversion of tumor-suppressor pathways. Because clonal hematopoiesis is not deterministic of malignant transformation, rational surveillance strategies will depend on the ability to prospectively identify specific clones with increased leukemic potential. We describe a framework by which an understanding of the processes that promote clonal hematopoiesis in IBMFSs may inform clinical surveillance strategies.
Introduction
Patients with inherited bone marrow failure syndromes (IBMFSs) have an elevated risk for developing myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) in the context of germline genetic alterations that compromise hematologic function. Myeloid transformation in IBMFSs has variable penetrance and latency but occurs at a younger age than in patients with sporadic myeloid malignancies.1 Allogeneic hematopoietic stem cell transplantation is the only potentially curative treatment, particularly when used prior to development of frank leukemia, but it carries a risk for transplant-related morbidity and mortality. Thus, a central clinical challenge is to predict in which patients and at what time leukemic transformation may occur.
Clinical surveillance strategies rely on monitoring hematologic status through serial measurement of peripheral blood counts and bone marrow biopsy examination, with the goal of identifying evidence of incipient transformation. Development of persistent severe cytopenias, morphologic dysplasia, or clonal cytogenetic alterations raises suspicion of progression toward MDS/AML. More recently, somatic point mutations in myeloid driver genes have been identified in patients with IBMFSs who do not have leukemia,2,3 raising the possibility that the presence of clonal hematopoiesis (CH) may be predictive of transformation. Here, we will review the processes that promote CH in the context of inherited bone marrow failure syndromes and discuss how an enhanced understanding of these processes will be required as a basis for development of rational surveillance strategies.
IBMFSs
The IBMFSs are unified by the presence of ineffective hematopoiesis that is due to germline errors in cellular homeostasis. Their pathophysiologies are variable and are linked to defects in diverse biological processes, including DNA damage repair (Fanconi anemia), telomere maintenance (dyskeratosis congenital [DC]), and ribosome biogenesis (Diamond-Blackfan anemia [DBA], Shwachman-Diamond syndrome [SDS]).4 The phenotypic spectrum of IBMFSs is similarly diverse, with marked differences in the number and severity of affected blood lineages, as well as variability in the magnitude of MDS and leukemia risk and the latency of leukemic transformation.1 Some, like Fanconi anemia and DC, display global stem cell defects and have a markedly elevated risk for developing myeloid malignancies early in life. Others, such as DBA, have a severe lineage-restricted hematologic marrow failure phenotype and a modestly increased leukemia risk.1 Germline mutations in other genes, including GATA2, CEBPA, RUNX1, SAMD9, SAMD9L, and DDX41, cause familial predisposition to myeloid malignancies without the characteristic features of classical IBMFSs.5-7
Normal and clonal hematopoiesis
Peripheral blood is maintained throughout the lifespan of an individual by bone marrow hematopoietic stem cells (HSCs), which are defined by their capacities of sustained self-renewal and multilineage differentiation.8 Under steady-state conditions, a diverse pool of HSC clones is maintained in homeostatic balance and contributes to polyclonal hematopoiesis. During normal aging, somatic genome diversification in individual HSCs occurs at a low, but detectable, frequency and is propagated during HSC self-renewal. In normal HSCs, somatic mutations have an age-associated signature that consists of C>T transitions within NCG trinucleotide sequences resulting from spontaneous deamination of 5-methylcytosine to thymine.9 These base substitutions accumulate in HSC genomes at a rate ∼14 mutations per year, including exonic mutations at a rate ∼1 every 10 years.9,10 Thus, steady-state polyclonal hematopoiesis reflects a balanced constituency of many individual clones, each defined in theory by a distinct complement of somatic genetic alterations.
CH is defined by the imbalanced contribution of a single HSC clone to the peripheral blood and can arise by neutral drift or directional selection. With neutral drift, all clones have the same chance of contributing to the pool of self-renewing HSCs, but random processes, such as stem cell attrition, lead to the fixation of some clones and loss of others. With positive selection, somatic alterations mediate selective growth advantage of specific HSC clones relative to others, leading to clonal dominance.
When identified based on the presence of leukemia-associated mutations in individuals with normal peripheral blood counts and no morphologic evidence of hematological malignancy, CH has been termed “clonal hematopoiesis of indeterminate potential" (CHIP).11 The presence of CHIP is associated with an increased risk for developing hematologic malignancy, with a rate of progression of ∼0.5% per year.12,13 The magnitude of risk is affected by clinical and genetic variables, such as the presence of concurrent cytopenias, increased clone size, and mutations in MDS-associated genes, each of which is associated with a greater risk for transformation.14-17
The link between CH and malignant disease has been described in the context of sporadic and therapy-related myeloid neoplasms.3,12,13,15,18-20 However, the impact of germline alterations on the likelihood of CH progression to frank malignancy has not been clearly defined. What are the rules that govern the development of somatic clones in IBMFSs? Does neutral drift due to HSC attrition underlie the general development of clonal mosaicism in IBMFSs, independent of underlying mechanisms? Does the germline lesion in a particular IBMFS drive disease-specific pathways of somatic clonal competition?
In this review, we will describe the mechanisms by which acquired somatic alterations can relieve germline fitness constraints in IBMFSs. Somatic normalization, via reversion or compensation, mediates clonal expansion by repairing germline-encoded cellular defects. "Reversion" occurs when the germline defect is directly corrected via gene conversion or back mutation or is inactivated by deletion or point mutation. "Compensation" occurs when the germline functional deficit is attenuated indirectly by somatic alteration of other genes, reducing cellular impact of the causative mutation. In contrast, somatic transformation occurs when mutations bypass the underlying germline defect by disrupting cellular tumor-suppressor or cellular fitness-sensing pathways.
Cell competition
The competitive fitness of a cell cannot be expressed in absolute terms, but is instead defined in specific context. Even severe functional defects in core biological pathways may have a modest impact on development and tissue homeostasis, unless occurring in direct juxtaposition to more competent cells. For example, in Drosophila wing development, cells with haploinsufficiency of the ribosomal protein-encoding gene Minute are capable of organogenesis in a noncompetitive environment. However, in a mosaic context with wild-type cells, Minute cells are actively eliminated, with preferential contribution of the more fit wild-type cells.21,22 This process of cell selection, termed “cell competition,” eliminates unfit clones that are generated by somatic genetic diversification during development.23,24
Cell competition can also promote tissue homeostasis during adulthood by protecting against deleterious genetic events or environmental stressors. When these changes affect baseline fitness within a tissue compartment, cell competition can mediate the expansion of somatic clones to maintain homeostatic balance. For example, in patients with ulcerative colitis, in which chronic inflammation of the intestinal tract leads to repeated cycles of epithelial destruction and regeneration, somatic mutations in genes involved in interleukin-17 (IL-17) signaling, such as NFKBIZ, were identified in clonally expanded intestinal crypt cells. These mutations were shown to confer resistance to IL-17–induced apoptosis, suggesting that clones with impaired NFKBIZ activity have a selective survival advantage in the setting of chronic inflammatory injury.25,26 Somatic mutations in NFKBIZ and related genes were not identified in biopsy samples from patients with colorectal cancer, indicating that acquired resistance to IL-17–dependent apoptosis reflects a specific adaptation to injury rather than a common feature of colorectal tumorigenesis.25 Similarly, PPM1D mutations are enriched in therapy-related CH, MDS, and AML as a result of selective advantage in the setting of cytotoxic chemotherapy, but they do not confer a competitive advantage in mouse serial transplantation models27 and do not regularly persist after autologous transplantation in humans.28 In these examples, cell competition maintains tissue homeostasis in the setting of chronic injury by favoring the contribution of clones with increased functional competency, without driving oncogenesis. These studies indicate that wild-type cells do not necessarily have maximal fitness and that they can be outcompeted by somatic clones that gain selective advantage in specific tissue or environmental contexts.
The acquired capacity to eliminate or outcompete wild-type cells is also a property of incipient cancer. In Drosophila, subversion of tumor-suppressor pathways or cooptation of oncogenic pathways can convert “loser” cells to “winner” cells, thereby enabling their preferential persistence despite functional impairment. This process, termed “supercompetition,” was observed in clones overexpressing Myc, which triggered active elimination of wild-type cells by a non–cell autonomous mechanism.29-31 A similar finding has been described in mouse models of hematopoiesis, where distinct mechanisms of sensing absolute and relative cell fitness rely on different facets of p53 pathway activity.32 Absolute fitness is acutely measured and defines a cell autonomous threshold of stress, such as DNA damage, that is tolerable for cell survival. Relative fitness is chronically surveyed among a group of cells with variable fitness and enforced via non–cell autonomous mechanisms. The convergence of absolute and relative fitness sensing on p53 activity suggests a mechanism by which genetic inactivation of cell-competition pathways favors selective growth of clones with enhanced oncogenic potential. An improved understanding of the non–cell autonomous pathways of cell competition in IBMFSs, as well as how they may be subverted to drive supercompetition or evasion of cell autonomous fitness constraints, may help to explain the mechanistic basis of leukemia predisposition.
Cell competition defines selection baseline
Laboratory models of IBMFSs suggest that the molecular pathogenesis of each disease defines a qualitatively distinct functional state at baseline. This baseline state may, in turn, define a distinct fitness set point for mosaic cell competition.
In a mouse model of germline telomerase deficiency, mutant HSCs were shown to have impaired proliferative potential and reduced colony-forming capacity.33 In the setting of noncompetitive transplantation, these defective HSCs were able to engraft and reconstitute multilineage hematopoiesis. However, when the same cells were transplanted along with wild-type competitors, reconstitution in recipient mice occurred preferentially from the wild-type cells.34 Similarly, mouse models of Fanconi anemia, including Fancc, Fancg, Fancd1, Fancd2, Fanci, and Usp1, have normal peripheral blood counts at steady-state,35-41 but yet defects in hematopoietic stem and progenitor cell (HSPC) function are highlighted in direct competition with wild-type or exogenously complemented cells, where Fanc-deficient cells are outcompeted.42 In Gata2-haploinsufficient mice, HSPCs were less abundant compared with Gata2 wild-type mice and competed poorly with wild-type HSPCs in competitive transplantation.43 In these models, telomerase-deficient, Fanc-deficient, or Gata2-haploinsufficient cells retained functional competence in a homogenous environment but were selectively eliminated as “losers” when surrounded by wild-type “winner” cells with enhanced fitness.
In contrast, mouse models of DBA indicate that ribosomal protein mutations specifically impair erythropoiesis via p53-induced cell cycle arrest, without having a significant impact on HSC function. Mice with Rpl11 haploinsufficiency showed a severe and selective defect in erythropoiesis, with markedly decreased numbers of erythroblasts but overall normal proportions of HSPCs.44 Mice with Rps14 haploinsufficiency showed a similar progressive anemia and no competitive disadvantage in the context of mixed marrow chimeras with wild-type cells.45 These data indicate that germline alterations can drive severe lineage-specific hematologic phenotypes without altering the function or competitive fitness of the stem cell compartment.
In these model systems, disease-specific fitness deficits can be directly assessed relative to wild-type cells. Further, these deficits can be characterized with stage and lineage specificity and interrogated for context dependence. Based on the available data, we hypothesize that clonal expansions harboring somatic genetic lesions that repair, compensate, or bypass germline encoded cellular vulnerabilities will manifest selectively in conditions such as telomere biology disorders and Fanconi anemia, where HSCs are “losers” in direct competition with wild-type “winners.” By contrast, the stage-specific phenotype in DBA models and the demonstrated lack of cell competition at the HSC level suggest that DBA HSCs do not have severe fitness constraints that drive potent selection of somatic clones capable of long-term persistence.
CH in bone marrow failure syndromes
DC
Telomere biology disorders are caused by germline mutations that impair telomere length maintenance. They comprise a heterogeneous set of patients with incompletely penetrant clinical phenotypes, most commonly aplastic anemia, idiopathic pulmonary and hepatic fibrosis, and early onset of hematologic and solid tumor malignancies. In DC, a severe early-onset syndromic presentation of telomere biology disorder, the risk of developing MDS (ratio of observed/expected, 578; 95% confidence interval, 343-918) or AML (ratio of observed/expected, 73; 95% confidence interval, 23-169) is markedly greater than in the general population.1
CH is common in individuals with telomere biology disorders.46 However, the spectrum of genetic alterations that define somatic clonal expansion in this context is remarkably different from what is reported in sporadic CH. Although sporadic CH is most commonly associated with mutations in DNMT3A, TET2, ASXL1, or other drivers of hematologic malignancies, these myeloid driver mutations are rare in DC.46,47 Instead, CH in DC has most often been described to occur without driver mutations, identified by the presence of skewed X chromosome inactivation (in females), somatic copy number alterations, or nonrecurrent somatic point mutations.46 This could represent neutral drift in the setting of HSC attrition or unrecognized compensatory events.
The most commonly identified cause of CH in the setting of telomere biology disorders is somatic reversion of, or compensation for, the pathogenic germline allele. This was first observed with recurrent tissue-restricted somatic mosaicism that resulted from mitotic gene conversion replacing a pathogenic mutated TERC allele with the wild-type TERC allele. This phenomenon was limited to hematopoietic cells and was identified in 6 individuals from 4 families.48 A similar somatic reversion event in an expanded hematopoietic clone was observed in a patient with germline DKC1 mutation.46 In a cohort of 199 patients with germline TERT mutations, 5% were found to have CH marked by somatic mutations in the TERT promoter. These mutations were seen exclusively in trans to the mutated TERT allele, caused increased transcription of the unaffected wild-type TERT allele, and led to enhanced telomerase activity.49 These 2 mechanisms mediate a similar functional outcome: enhanced competitive fitness by somatic improvement of cellular telomerase activity. Although reversion accomplishes this effect by directly removing the mutated allele, compensation via TERT promoter mutations does so by augmenting expression of normal telomerase in the setting of an intact mutated allele.
The genetic pathways that drive leukemic transformation in telomere biology diseases are incompletely understood and may be different from the pathways that drive CH. In 1 registry-level study focused on MDS, patients with putative germline TERT or TERC mutations were found to have frequent somatic mutations in TP53 and PPM1D.50 TP53 and PPM1D are central effectors of the DNA damage response evoked by deprotected telomere ends,51-56 raising the possibility that telomere attrition within the HSC population selects for cells with genetic capacity to escape senescence pathways without needing to repair underlying defects in telomere lengthening.
SDS
SDS is an autosomal-recessive disease caused primarily by biallelic germline SBDS mutations. SBDS mutations are most often present in a compound heterozygous conformation with 1 null allele (nonsense mutation) and 1 hypomorphic allele (splice site mutation), where severe SBDS deficiency results in impaired ribosome maturation, translational inefficiency, and p53 activation leading to cellular senescence.57-59 SDS manifests clinically with skeletal abnormalities, pancreatic insufficiency, and bone marrow failure with high risk for transformation to MDS and AML.60
CH associated with 2 recurrent cytogenetic alterations has been described in patients with SDS.61 In patients with compound heterozygous SBDS mutations, recurrent acquisition of isochrome 7q [i(7)(q10)] has been observed in hematopoietic cells. The i(7q) encompasses the SBDS locus and, thus, duplicates a hypomorphic SBDS splice site allele, presumably increasing the absolute level of cellular SBDS. Interstitial deletion of 20q is observed in ∼20% of patients with SDS and is associated with benign clinical outcomes.62,63 The common deleted region on 20q encompasses ≥68 genes,64,65 but unlike in del(5q) MDS,45,66,67 no systematic functional analyses have definitively assigned a causal link between haploinsufficiency of a single gene and development of clonal advantage.
EIF6 haploinsufficiency has been proposed to contribute to the selective clonal advantage conferred by del(20q) in SDS hematopoiesis. EIF6 is localized to the SDS del(20q) common deleted region and encodes an essential ribosome chaperone protein that binds to the nascent 60S ribosome subunit in the nucleolus. Upon export to the cytoplasm, EIF6 release from the nascent 60S ribosome is catalyzed by SBDS/EFL1, thereby allowing subunit joining and activation of translation.68-71 Reduced EIF6 levels have been shown to improve ribosomal subunit association in SDS patient–derived bone marrow stromal cells,70 suggesting that EIF6 haploinsufficiency attenuates the germline defect through functional normalization of ribosome maturation.
A recent study from the North American Shwachman-Diamond Syndrome Registry identified highly recurrent somatic EIF6 mutations in bone marrow samples from SDS patients but not from patients with other IBMFSs or predisposition syndromes (DC, GATA2 deficiency syndrome, or germline SAMD9/SAMD9L mutations) or sporadic AML.72 In functional analyses, these EIF6 mutations were shown to disrupt EIF6:60S binding interaction or destabilize EIF6 protein, resulting in improved 80S maturation and global protein synthesis and a reduction in aberrant p53 activation. Overall, somatic clones were infrequent in the first several years of life but approached ubiquity in the second decade and beyond, and most commonly involved 1 of only 4 genes (EIF6, TP53, PRPF8, and CSNK1A1).
Together, the recurrence of somatic EIF6 mutations and i(7q) and del(20q) alterations suggests that normalizing the ratio of SBDS:EIF6 in SDS hematopoietic cells, by augmenting SBDS levels or reducing EIF6 levels, enhances competitive fitness by improving ribosome maturation and translational capacity. These alterations are not associated with leukemic transformation, further suggesting that functional correction of germline-encoded cellular defects drives enhanced fitness of somatic clones without altering normal pathways of oncogene protection.
In a cohort of MDS patients who underwent hematopoietic stem cell transplantation, all 7 patients with genetically defined SDS (biallelic SBDS mutations) had ≥1 TP53 mutation, suggesting that somatic TP53 mutations mediate myeloid transformation in SDS.50 However, a subsequent analysis of patients with congenital neutropenia syndromes identified ≥1 TP53 mutation in 13 of 27 patients with SDS but found that the presence of TP53-mutated CH was not associated with specific hematologic manifestations, including peripheral blood counts or myeloid neoplasms.73 In the SDS Registry study, the presence, number, and allele abundance of somatic TP53 mutations were not predictive of imminent leupi kemia risk in SDS patients. Instead, progression of TP53-mutated clones was mediated by the development of biallelic alterations in the TP53 locus, by deletion, copy neutral loss of heterozygosity, or second point mutation.72 In functional experiments, TP53 inactivation was shown to enhance the competitive fitness of SBDS-deficient cells by inactivating senescence pathways without correcting the underlying SDS ribosome and translational defects. Consistent with these human genetic observations, loss of TP53 in a mouse model of SDS can uncouple the ribosomal stress caused by SBDS deficiency from activation of cellular senescence pathways, resulting in partial rescue of the SDS phenotype.58,59
Fanconi anemia
Fanconi anemia is caused by germline mutations in the FANC cluster of genes that coordinate DNA damage repair by homologous recombination. Patients with Fanconi anemia have increased sensitivity to DNA-damaging agents and manifest clinically with congenital developmental defects, bone marrow failure, and an elevated risk for developing MDS and AML that is several hundred–fold higher than in unaffected individuals.74,75 Somatic mosaicism was recognized in the blood of patients with Fanconi anemia who had milder cytopenias and improved blood counts. In those with compound heterozygous germline Fanconi mutations, reversion has been suggested to occur via gene conversion or back mutation.76,77 Similarly, patients with homozygous Fanconi mutations, where gene conversion would not be available as a mechanism of reversion, were found to have clonal populations of circulating lymphocytes with somatic missense and frameshift mutations in cis with the germline FANC allele.78 In vitro analyses of these secondarily mutated genes revealed that the somatic changes resulted in a gene product with restored wild-type function and improved DNA damage repair capability.76-80 The propensity for somatic reversion of the germline Fanconi genotype and the diversity of molecular mechanisms suggest that there is a strong selective advantage to eliminate the HSC fitness constraint in Fanconi anemia.
CH in leukemia-predisposition syndromes not associated with IBMFS
The mechanisms underlying development of CH in the classical IBMFSs may not apply to all leukemia-predisposition syndromes. For example, somatic normalization has not been observed in patients with germline mutations in RUNX1, CEBPA, DDX41, or GATA2. Instead, somatic mutations in CH and myeloid neoplasms in these contexts have been reported to arise as a second hit in the unaffected allele of the germline mutated gene (as seen in RUNX1, CEBPA, and DDX41)6,81-83 or as secondary mutations in typical myeloid driver genes (as seen in GATA2 and RUNX1).3,84,85 This may reflect an absence of germline-encoded HSC fitness constraint in these contexts or the limited genomic characterization of samples from patients with these diseases.
In contrast, somatic reversion leading to CH has been observed in patients with germline gain-of-function mutations in SAMD9/SAMD9L, which are associated with MDS predisposition.7,86-88 Acquired deletion of chromosome 7q at the locus of the SAMD9/SAMD9L mutation is a common event, causing haploinsufficiency that improves HSC survival and clinically leads to milder cytopenias, although with an increased risk for myeloid transformation.7,86-90 Somatic mosaicism was observed in 5 individuals from 2 families with mitotic recombination leading to uniparental disomy of chromosome 7q and copy number–neutral loss of heterozygosity of the germline SAMD9L mutation.87 Two carriers also acquired a revertant truncating mutation within the germline mutated SAMD9L allele, representing a loss-of-function rescue.87 Similarly, copy number–neutral loss of heterozygosity of chromosome 1p was observed to be strongly associated with inherited rare variants of MPL in otherwise healthy individuals, with the preferential loss of the mutant MPL allele suggesting a selective advantage to restoring normal MPL function.91
Conclusions
In IBMFSs, HSC fitness constraints define a globally altered baseline (Figure 1). When these baseline functional impairments are present in the self-renewing HSC compartment, somatic mutations can improve the competitive fitness of individual HSC clones by correcting or mitigating the germline fitness constraint. Any somatic mutation that improves or normalizes the lesional pathway through reversion or compensation may provoke the biological mechanism of cell competition within the HSC compartment. This enables selective expansion of more competent somatic clones and elimination of less competent baseline HSCs. Because normalization occurs without inactivating tumor-suppressor pathways, it does not confer an increased risk for transformation. Laboratory models of IBMFSs support this concept and may help to identify germline lesions that cause HSC-intrinsic fitness constraints through direct comparison of relative fitness in mixed bone marrow chimeras.
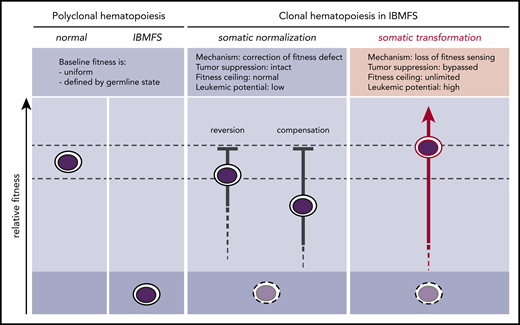
Somatic pathways of clonal expansion in IBMFSs. HSPCs in IBMFSs have impaired baseline global fitness compared with normal HSPCs. Somatic mutations can lead to the development of CH by improving fitness over the germline state. Somatic normalization occurs through correction of the fitness defect by reversion or compensation, with intact tumor-suppressor pathways. Improvement in fitness is limited to a maximum of wild-type levels, and leukemic potential is low. In contrast, somatic transformation occurs through loss of fitness sensing, with bypass of tumor-suppressor pathways. Improvement in fitness is theoretically unlimited, and leukemic potential is high.
Somatic pathways of clonal expansion in IBMFSs. HSPCs in IBMFSs have impaired baseline global fitness compared with normal HSPCs. Somatic mutations can lead to the development of CH by improving fitness over the germline state. Somatic normalization occurs through correction of the fitness defect by reversion or compensation, with intact tumor-suppressor pathways. Improvement in fitness is limited to a maximum of wild-type levels, and leukemic potential is low. In contrast, somatic transformation occurs through loss of fitness sensing, with bypass of tumor-suppressor pathways. Improvement in fitness is theoretically unlimited, and leukemic potential is high.
Other somatic mutations bypass tumor-suppressor pathways without correcting the underlying germline defect. In animal models of SDS and DC, for example, inactivation of TP53 limits the cellular consequences of translational inefficiency and defective telomere length, respectively, indicating that p53 pathway activation limits relative fitness and mediates elimination of “losers.” Thus, inactivation of TP53 in single HSC clones could convert “losers” to “winners,” consistent with the observed recurrence of somatic TP53 mutations in CH and myeloid neoplasms that arise in SDS and telomere biology disorders.
Clinical implications
CH itself does not indicate impending myeloid transformation. Thus, rational surveillance strategies depend on the ability to prospectively identify specific clones with increased leukemic potential (Figure 2). In theory, this could be accomplished by discriminating expanded clones driven by alterations that directly subvert fitness-sensing and tumor-suppressor pathways from those that cause somatic normalization (through reversion and compensation). Although clones with somatic normalization may have limited transformation potential because of the retention of functionally intact fitness-sensing pathways, those with maladaptive mutations that impair cellular elimination may have an increased risk for leukemic transformation that is due to subversion of these pathways. It is not known whether leukemia risk is clone autonomous or whether other microenvironmental or patient-specific factors contribute. Systematic analysis of registry-level cohorts with adequate clinical annotation and duration of follow-up will be required to further derive and validate more general and disease-specific principles. Prospective clinical studies will further assist in the integration of rational surveillance strategies with clinical care aimed at optimizing the timing of transplantation or deployment of novel leukemia interventions.
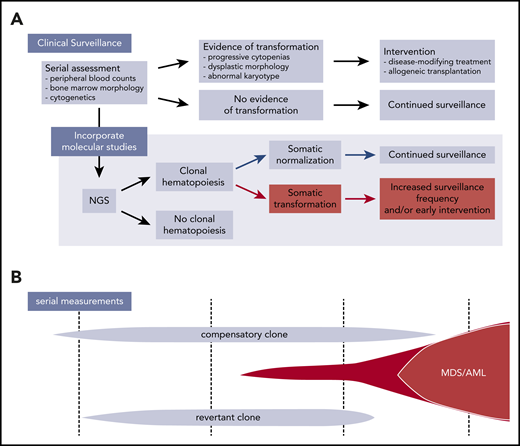
Incorporation of next-generation sequencing (NGS) into surveillance in IBMFS patients. (A) Current surveillance strategies in IBMFSs rely on serial monitoring of peripheral blood counts, bone marrow morphology, and cytogenetics. Incorporation of NGS to identify CH and assign specific mechanisms of somatic mutation may allow for early detection of clones with increased transformation risk and enable early intervention. (B) Schematic of CH in a patient with IBMFS. Distinct clones can coexist over time, with only certain transformation-associated clones at risk for evolving into MDS/AML.
Incorporation of next-generation sequencing (NGS) into surveillance in IBMFS patients. (A) Current surveillance strategies in IBMFSs rely on serial monitoring of peripheral blood counts, bone marrow morphology, and cytogenetics. Incorporation of NGS to identify CH and assign specific mechanisms of somatic mutation may allow for early detection of clones with increased transformation risk and enable early intervention. (B) Schematic of CH in a patient with IBMFS. Distinct clones can coexist over time, with only certain transformation-associated clones at risk for evolving into MDS/AML.
Acknowledgment
This work was supported by National Institutes of Health, National Cancer Institute grants T32 CA009172 and K08 CA204734.
Authorship
Contribution: F.D.T. and R.C.L. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: R. Coleman Lindsley, Dana-Farber Cancer Institute, 450 Brookline Ave – DA-530C, Boston, MA 02215; e-mail: coleman_lindsley@dfci.harvard.edu.
