Key Points
SARS-CoV-2 spike proteins bind heparan sulfate and activate the alternative complement pathway on cell surfaces.
Factor D inhibitor (ACH145951) blocks the complement activation induced by SARS-CoV-2 spike proteins.

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a highly contagious respiratory virus that can lead to venous/arterial thrombosis, stroke, renal failure, myocardial infarction, thrombocytopenia, and other end-organ damage. Animal models demonstrating end-organ protection in C3-deficient mice and evidence of complement activation in humans have led to the hypothesis that SARS-CoV-2 triggers complement-mediated endothelial damage, but the mechanism is unclear. Here, we demonstrate that the SARS-CoV-2 spike protein (subunit 1 and 2), but not the N protein, directly activates the alternative pathway of complement (APC). Complement-dependent killing using the modified Ham test is blocked by either C5 or factor D inhibition. C3 fragments and C5b-9 are deposited on TF1PIGAnull target cells, and complement factor Bb is increased in the supernatant from spike protein–treated cells. C5 inhibition prevents the accumulation of C5b-9 on cells, but not C3c; however, factor D inhibition prevents both C3c and C5b-9 accumulation. Addition of factor H mitigates the complement attack. In conclusion, SARS-CoV-2 spike proteins convert nonactivator surfaces to activator surfaces by preventing the inactivation of the cell-surface APC convertase. APC activation may explain many of the clinical manifestations (microangiopathy, thrombocytopenia, renal injury, and thrombophilia) of COVID-19 that are also observed in other complement-driven diseases such as atypical hemolytic uremic syndrome and catastrophic antiphospholipid antibody syndrome. C5 inhibition prevents accumulation of C5b-9 in vitro but does not prevent upstream complement activation in response to SARS-CoV-2 spike proteins.
Introduction
COVID-19, the disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is causing an unprecedented global pandemic, resulting in nearly 1 million deaths since late 2019. Coronaviruses (CoVs) are single-stranded, positive-sense, RNA viruses first identified over 70 years ago.1 CoVs received considerable attention in 2003 when the etiologic agent of severe adult respiratory syndrome (SARS-CoV) was identified as a CoV.2,3 The CoV genome encodes for 4 main structural proteins, spike (S), membrane (M), envelope (E), nucleocapsid (N), as well as other accessory proteins that facilitate replication and entry into cells.4 The spike proteins, consisting of S1 and S2 subunits, cover the surface of CoVs and serve as entry proteins for infection.4 Spike proteins first attach to the cells by binding to glycosaminoglycans such as heparan sulfate (HS) and α2,3 and α2,6 sialylated N-glycans, which further facilitate the interaction with entry receptors and membrane fusion with host cells.5,6 SARS-CoV-2 uses the angiotensin-converting enzyme-2 as its entry receptor, which is widely expressed on lung type II alveolar cells, enterocytes, arterial and venous endothelial cells, and arterial smooth muscle cells in most organs.7
COVID-19 is highly contagious and spreads predominantly by respiratory droplets.8 Clinical manifestations are manifold, ranging from asymptomatic infection to multiorgan failure and death in 2% to 10% of patients.9 Most symptomatic patients complain of a flu-like illness consisting of fever, cough, nasal congestion, fatigue, and myalgia. A small percentage of patients develop more severe symptoms affecting multiple organ systems. Refractory thrombosis,10 thrombotic microangiopathies,11,12 antiphospholipid antibodies,13,14 stroke,15 seizures, respiratory failure,16 renal failure,17 and myocardial infarction are well-described complications of COVID-19 but occur in a minority of patients. It remains unclear why only a small subset of SARS-CoV-2–infected patients acquires such severe endothelial damage affecting multiple organs.
The recalcitrant hypercoagulability, thrombotic microangiopathy, diffuse endothelial damage, and the inflammatory milieu associated with severe COVID-19 infection have led to the hypothesis that excessive complement activation may be responsible for the end-organ damage, but the mechanism is unclear.12,18,19 Indeed, complementopathies, such as atypical hemolytic uremic syndrome (aHUS) and catastrophic antiphospholipid antibody syndrome (CAPS), share many clinical features with severe COVID-19 infection.20,21
Preclinical data have also demonstrated a role for complement activation in CoV-mediated disease. Gralinski et al evaluated the activation of the complement system in a mouse model of CoV.22 Relative to C57BL/6J control mice, SARS-CoV–infected C3−/− mice exhibited significantly less weight loss and less respiratory dysfunction despite equivalent viral loads in the lung. Transgenic animals lacking C3 also had reduced inflammatory cells in the large airway and parenchyma, improved respiratory function, and lower levels of inflammatory cytokines or chemokines in the lung and periphery. Middle Eastern respiratory syndrome–CoV infection in mice have severe acute respiratory failure and high mortality accompanied by elevated secretion of cytokines and chemokines. Increased concentrations of C5a and C5b-9 (terminal complement complex), activation products resulting from cleavage of C5, were detected in sera and lung tissue in the infected mice, respectively.23
In humans with COVID-19 pneumonia, C5b-9, C4d, and mannan-binding lectin serine protease 2 are found in the microvasculature of the lung, and COVID-19–associated skin lesions exhibit colocalization of SARS-CoV-2 spike proteins with C4d and C5b-9 in the cutaneous microvasculature.24 A prospective cohort study of 150 patients with COVID-19 acute respiratory distress syndrome found a high incidence of pulmonary emboli (17%) despite prophylactic anticoagulation.25 Here, we show that the SARS-CoV-2 spike protein subunits, but not N protein or spike protein from a more benign human CoV (OC43), are potent activators of the alternative pathway of complement (APC), and that C5 and factor D inhibitors prevent complement-mediated damage.
Materials and methods
Human CoV proteins
Recombinant proteins expressed with the Escherichia coli system include SARS-CoV-2 S1 subunit protein (receptor-binding domain [RBD]) (S1; RayBiotech), S2 subunit (S2; RayBiotech), and nucleocapsid protein (N; ABclonal Technology). Recombinant proteins expressed with the baculovirus-insect cell system include human CoV spike protein (HCoV-OC43 S; Sino Biological). CoV proteins were used to activate complement by adding into normal human serum (NHS; Complement Technology, Inc). Heat denaturation of human CoV proteins was performed by heating proteins at 100°C for 30 minutes.
The modified Ham test
The modified Ham (mHam) assay was used to test complement activation in serum as described previously.26
Cell preparation
TF1PIGAnull cells were maintained at a density of 500 000 cells per milliliter daily.26 Before the assay, the cells were washed with phosphate-buffered saline (PBS) and seeded in a round-bottom 96-well plate at a density of 6700 cells per well in 80 µL of gelatin veronal buffer with Ca and Mg (GVB++, Complement Technology, Inc) in triplicate.
Serum preparation
NHS (20 µL; Complement Technology, Inc) was added with 0.25, 0.5, 1.0, and 2.0 µg of human CoV proteins (final concentrations of S1, S2, N, and HCoV-OC43 S are 2.5, 5, 10, and 20 µg/mL) and incubated on ice for 15 minutes. As a negative control, NHS was heated at 56°C for 30 minutes to inactivate NHS complement activity (NHS [H]). For the complement rescuing mHam, NHS was first incubated with either a small molecule factor D inhibitor27 (ACH145951; Achillion Pharmaceuticals) diluted in dimethyl sulfoxide (final concentration, 1.0 µM) or 50 µg of anti-C5 monoclonal antibody (anti-C5Ab; Alexion Pharmaceuticals) on ice for 15 minutes, and then added with SARS-CoV-2 spike proteins for another 15 minutes on ice.
Complement reaction
The 20-µL serum mixture was added to 80 µL cells and incubated at 37°C for 45 minutes with constant shaking. After incubation, cells were separated by centrifugation at 600g for 3 minutes at room temperature and washed with PBS.
Cell viability assay
After washing, the cells in each well were resuspended in 100 µL of 10% WST-1 proliferation solution (WST-1: RPMI 1640 without phenol red at a ratio of 1:9, WST-1; Roche) and incubated for 2 hours at 37°C. WST-1 solution only was used as a blank control. The absorbance of the chromogenic metabolized product was measured with a plate reader (ELX808; BioTeK) at 450 nm with a reference wavelength at 630 nm.
Percentage of nonviable cell calculation
The sample absorbance value was normalized by subtracting the absorbance of a blank control. The percentage of live cells was calculated as the ratio of normalized sample absorbance (A450-630nm) to normalized negative control NHS(H) absorbance multiplied by 100 (formula: % live cells = [(sample − blank) / (NHS (H) − blank) × 100]). Complement activation level was indicated by the percentage of nonviable cells (100 − % live cells). Based on a receiver operative curve, ≥20% nonviable cells (cell killing) have been established as a positive test.28
Detection of complement activity by flow cytometry
Cell-surface depositions of C5b-9, C3c, and C4d on TF1PIGAnull cells were measured by flow cytometry.
Cell preparation
Before the assay, TF1PIGAnull cells were washed with PBS and seeded in V-bottom 96-well plates (1.2 × 105 cells per well) in 80 µL of either GVB++ buffer or GVB0 10 mM MgEGTA buffer (pH 6.4) (GVB0; Complement Technology, Inc). GVB++ allows all complement pathway activation whereas GVB0 MgEGTA only allows alternative pathway activation.
Serum preparation
NHS (20 µL) was added with 0.25, 0.5, 1.0, and 2.0 µg of human CoV proteins (final concentration of S1, S2, N, and HCoV-OC43 S ranges from 2.5 to 20 µg/mL) and incubated on ice for 15 minutes. For alternative pathway activation, NHS was acidified to pH 6.4 by adding 0.2 M HCl.
Complement reaction
The 80 µL cells were incubated with the 20-µL serum mixture for 15 minutes at 37°C with constant shaking, and the reaction was stopped by adding fluorescence-activated cell sorting buffer (PBS supplemented with 1% bovine serum albumin [BSA] and 15 mM EDTA). Cells were centrifuged at 600g for 3 minutes at room temperature and washed with PBS. NHS with 5 mM EDTA, which inhibits complement activation, was used as a negative control. As a positive control for C4d detection, 10 µg/mL Shiga toxin 1 (Sigma-Aldrich) was incubated with NHS on ice for 15 minutes, followed by the addition of cells in GVB++ buffer. Complement inhibitors, ACH145951 (final concentration 1.0 µM) and anti-C5Ab (50 µg per sample), were also used to identify the specific complement pathway(s) involved.
Staining and detecting
Cells were washed with PBS and stained with anti-C5b-9 monoclonal antibody (Santa Cruz Biotechnology, Inc; dilution at 1/100) for 30 minutes on ice. Then, cells were washed with PBS and stained with Alexa 647–conjugated secondary antibody (1/500 dilution; Abcam) and Alexa 488–conjugated anti-C3c antibody (1/150 dilution; Abcam) for another 30 minutes on ice. The cells were also labeled with anti-C4d biotinylated monoclonal antibody (1/50 dilution; Quidel) and phycoerythrin-streptavidin (1/500 dilution; BD Pharmingen). Ten thousand events per sample were collected by a BD FACSCalibur and data were analyzed using FlowJo software version 10.5.3 (FlowJo Inc).
Quantification of serum factor Bb by enzyme-linked immunosorbent assay
Serum Bb level was measured by a MicroVue Bb Plus EIA kit (Quidel). To determine the increase of serum Bb concentration in the presence of cells, 20 µL of NHS was preincubated with 20 µg/mL SARS-CoV-2 spike proteins for 15 minutes on ice, followed by the addition of 80 µL of either GVB0 MgEGTA buffer (pH 6.4) or TF1PIGAnull cells (1.2 × 105 cells per sample) in GVB0 MgEGTA buffer (pH 6.4). After the reaction was incubated for 15 minutes at 37°C, the cells were centrifuged at 600g for 3 minutes at room temperature and the supernatant was collected for Bb quantification. We also performed the assay with ACH145951 (1.0 µM) and anti-C5Ab (50 µg per sample).
Flow cytometry assay for SARS-CoV-2 spike proteins binding to TF1PIGAnull cells and blockade with HS
Flow cytometry was performed to evaluate the binding of SARS-CoV-2 spike proteins onto the cell surface. Heparan sulfate sodium salt (NaHS; Sigma-Aldrich) was dissolved in PBS with 0.5% BSA to reach a concentration of 2 mg/mL. TF1PIGAnull cells (50 000 cells per sample) were washed and resuspended in either 100 µL of PBS with 0.5% BSA or the NaHS/PBS solution with 0.5% BSA. The cells were then added with 5 µg/mL S1 or S2 with C-terminal His-tag and incubated for 15 minutes at 37°C. After incubation, the cells were washed and fixed with 4% formaldehyde solution in PBS for 15 minutes at room temperature. To measure the binding of His-tagged S1 and S2 to the cell surface, the cells were stained with anti-His-tag antibody (1/100 dilution; Santa Cruz Biotechnology, Inc) for 30 minutes on ice, followed by Alexa 488 goat anti-mouse immunoglobulin G (Invitrogen, dilution at 1/200) for 10 minutes on ice in the dark. His-tagged S1 and S2 bindings to cells were measured by a BD FACSCalibur.
Complement inhibition with purified factor H protein
Purified factor H proteins (10 µg; Complement Technology, Inc) and 20 µg/mL S1 or S2 were added to 20 µL of NHS and incubated for 15 minutes at 37°C. TF1PIGAnull cells (120 000 cells per sample) in 80 µL of GVB0 MgEGTA buffer (pH 6.4) were then added to the serum mixture followed by incubation for 15 minutes at 37°C with constant shaking. After the reaction, the cells were washed and stained with an anti-C5b-9 monoclonal antibody (1/100 dilution; Santa Cruz Biotechnology, Inc) on ice for 30 minutes followed by Alexa 647–conjugated secondary antibody (1/500 dilution; Abcam). Cells were also stained with Alexa 488–conjugated anti-C3c antibody (1/150 dilution; Abcam). C5b-9 and C3c depositions on the cell surface were measured by a BD FACSCalibur.
Data analysis
All experiments were performed at least 3 times. All data were summarized as mean plus or minus standard deviation or standard error; the Student t test was used to assess the difference between unpaired groups. A value of P < .05 was considered statistically significant.
Results
SARS-CoV-2 spike proteins (S1 and S2) induce cell killing through the APC
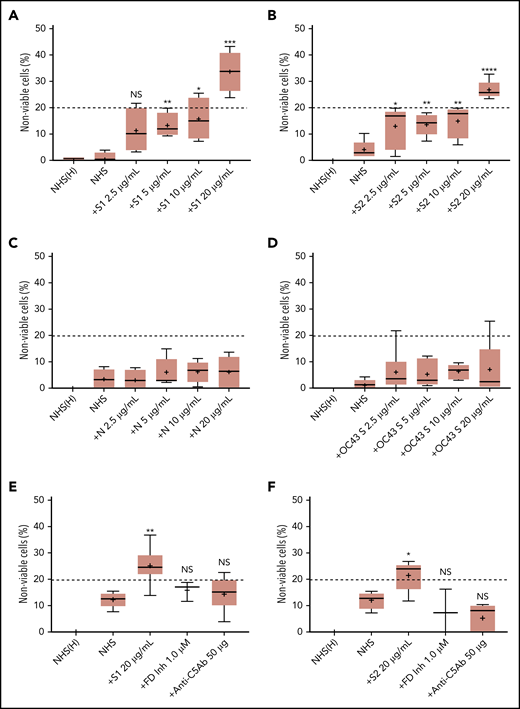
We first tested the ability of spike proteins to activate complement via a cell-based mHam assay that has been previously validated for detecting complement-driven diseases such as aHUS, CAPS, and the hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome.20,26,28 Spike protein S1 and S2 subunits from SARS-CoV-2 added to NHS induced dose-dependent cell killing in the mHam assay (Figure 1A-B), which was inhibited by a factor D inhibitor (ACH145951) and an anti-C5 monoclonal antibody (Figure 1E-F). The SARS-CoV-2 N protein and the spike protein from the benign human CoV OC43 did not increase killing in the mHam (Figure 1C-D). Because factor D is specific to the APC, these data suggest that the S1 and S2 subunits of SARS-CoV-2 spike protein activate complement primarily through the APC.
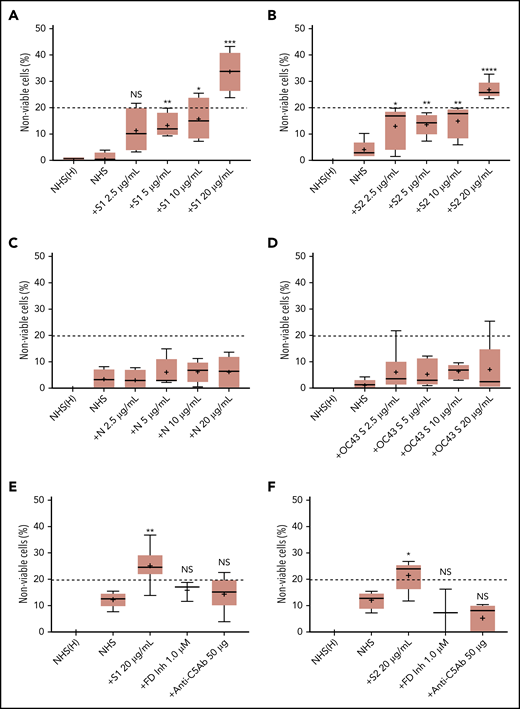
SARS-CoV-2 spike proteins induce complement-mediated cell killing that can be blocked by complement inhibitors in the mHam assay. TF1PIGAnull cells were treated with 20% NHS preincubated with diluted SARS-CoV-2 spike protein subunit 1 (S1), subunit 2 (S2), N proteins (N), and HCoV-OC43 S proteins (2.5 µg/mL to 20 µg/mL), and then measured for cell killing. Complement-mediated cell killing (%) was markedly increased in a dose-dependent manner with addition of S1 (A) and S2 (B). Increasing the concentration of N protein (C) and HCoV-OC43 S protein (D) did not increase the cell death (%) from baseline NHS level. Complement inhibition with 1 µM factor D inhibitor or 50 µg of anti-C5 monoclonal antibody completely blocked the cell killing induced by 20 µg/mL S1 (E) and S2 (F). The dotted line at 20% nonviable cells was established as a threshold for a positive mHam based on a receiver operative curve. All experiments were repeated at least 3 times. Statistical significance was calculated between each CoV protein–treated group and the NHS-treated group (*P < .05, **P < .01, ***P < .001, ****P < .0001). Anti-C5Ab indicates anti-C5 monoclonal antibody; FD Inh, factor D inhibitor (ACH145951); NHS(H), heat-inactivated NHS; NS, not significant.
SARS-CoV-2 spike proteins induce complement-mediated cell killing that can be blocked by complement inhibitors in the mHam assay. TF1PIGAnull cells were treated with 20% NHS preincubated with diluted SARS-CoV-2 spike protein subunit 1 (S1), subunit 2 (S2), N proteins (N), and HCoV-OC43 S proteins (2.5 µg/mL to 20 µg/mL), and then measured for cell killing. Complement-mediated cell killing (%) was markedly increased in a dose-dependent manner with addition of S1 (A) and S2 (B). Increasing the concentration of N protein (C) and HCoV-OC43 S protein (D) did not increase the cell death (%) from baseline NHS level. Complement inhibition with 1 µM factor D inhibitor or 50 µg of anti-C5 monoclonal antibody completely blocked the cell killing induced by 20 µg/mL S1 (E) and S2 (F). The dotted line at 20% nonviable cells was established as a threshold for a positive mHam based on a receiver operative curve. All experiments were repeated at least 3 times. Statistical significance was calculated between each CoV protein–treated group and the NHS-treated group (*P < .05, **P < .01, ***P < .001, ****P < .0001). Anti-C5Ab indicates anti-C5 monoclonal antibody; FD Inh, factor D inhibitor (ACH145951); NHS(H), heat-inactivated NHS; NS, not significant.
SARS-CoV-2 spike proteins (S1 and S2) increase APC markers on cells
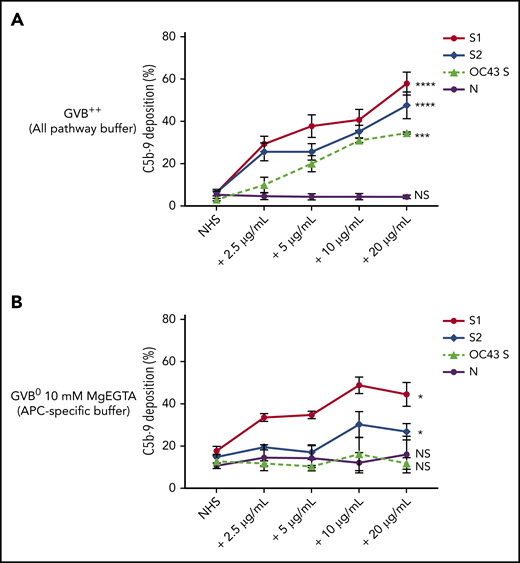
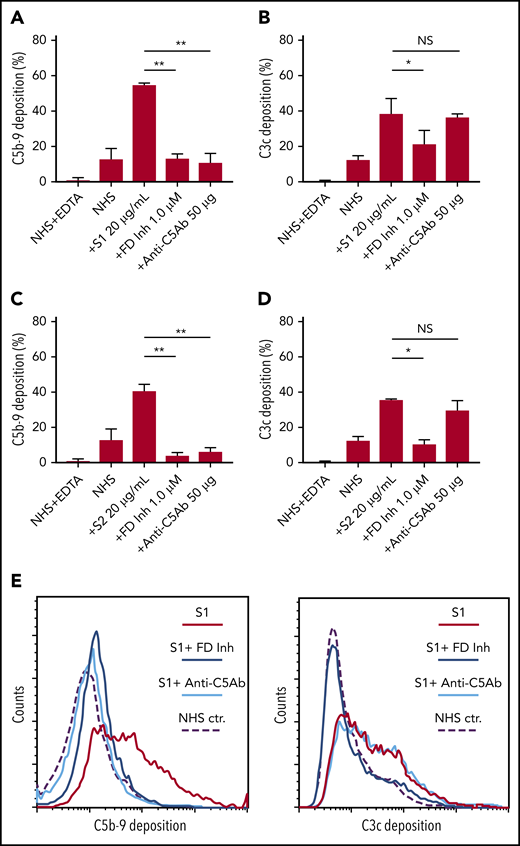
Biomarkers of complement activation, C3c, C4d, and C5b-9 deposition, were detected with flow cytometry. S1 and S2 subunits from SARS-CoV-2 added to NHS increased C5b-9 deposition in a dose-dependent manner that correlated with complement-dependent killing in the mHam (Figure 2). OC43 S protein only increased C5b-9 deposition in GVB++ buffer, indicating that it activated complement to a lesser degree through the classic and/or lectin pathways (Figure 2), which was not enough to reach the killing threshold for a nucleated cell (Figure 1D). Denaturing the S1 and S2 proteins with heat before adding to NHS significantly reduced the accumulation of C5b-9 deposition on the cell surface (supplemental Figure 1, available on the Blood Web site). C5b-9 deposition induced by S1 and S2 in both GVB++ and GVB0 MgEGTA buffer was inhibited by anti-C5Ab and ACH145951; however, ACH145951 was more potent in blocking C3c deposition because it targets the upstream cascade of the APC, especially in GVB0 MgEGTA (Figure 3). C4d deposition was modestly increased but did not show statistical significance (supplemental Figure 2). Taken together, these data confirm that S1 and S2 subunits of SARS-CoV-2 spike protein mainly activate the alternative pathway.
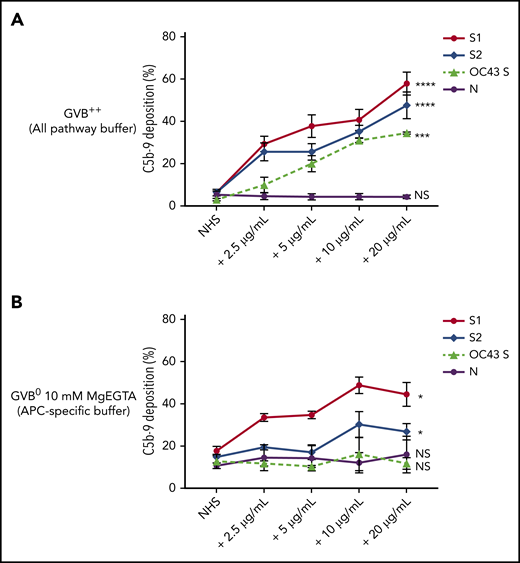
SARS-CoV-2 spike proteins induce C5b-9 deposition on the cell surface mainly through the alternative pathway. Flow cytometry demonstrated C5b-9 deposition on TF1PIGAnull cells after adding NHS preincubated with SARS-CoV-2 S1, S2, N, and HCoV-OC43 S proteins (2.5 µg/mL to 20 µg/mL) in either all pathway buffer (GVB++ pH 7.4) or APC-specific buffer (GVB0 10 mM MgEGTA pH 6.4). (A) SARS-CoV-2 S1, S2, and HCoV-OC43 S proteins elevated C5b-9 deposition in a dose-dependent manner in GVB++ buffer, whereas N protein did not increase C5b-9 from the baseline NHS level. (B) Both SARS-CoV-2 S1 and S2 led to marked increase of C5b-9 depositions in APC-specific buffer. By contrast, SARS-CoV-2 N and HCoV-OC43 S proteins showed minimal C5b-9 increase in APC-specific buffer. All experiments were repeated 6 times. Statistical significance was calculated between each 20 µg/mL CoV protein-treated group and the NHS-treated group (*P < .05, **P < .01, ***P < .001, ****P < .0001).
SARS-CoV-2 spike proteins induce C5b-9 deposition on the cell surface mainly through the alternative pathway. Flow cytometry demonstrated C5b-9 deposition on TF1PIGAnull cells after adding NHS preincubated with SARS-CoV-2 S1, S2, N, and HCoV-OC43 S proteins (2.5 µg/mL to 20 µg/mL) in either all pathway buffer (GVB++ pH 7.4) or APC-specific buffer (GVB0 10 mM MgEGTA pH 6.4). (A) SARS-CoV-2 S1, S2, and HCoV-OC43 S proteins elevated C5b-9 deposition in a dose-dependent manner in GVB++ buffer, whereas N protein did not increase C5b-9 from the baseline NHS level. (B) Both SARS-CoV-2 S1 and S2 led to marked increase of C5b-9 depositions in APC-specific buffer. By contrast, SARS-CoV-2 N and HCoV-OC43 S proteins showed minimal C5b-9 increase in APC-specific buffer. All experiments were repeated 6 times. Statistical significance was calculated between each 20 µg/mL CoV protein-treated group and the NHS-treated group (*P < .05, **P < .01, ***P < .001, ****P < .0001).
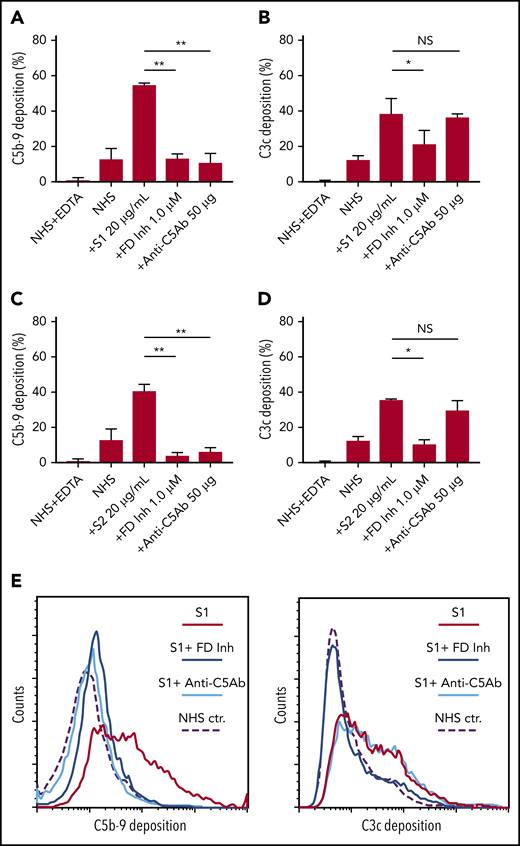
C5 and factor D inhibition block complement activation induced by SARS-CoV-2 spike proteins. Flow cytometry demonstrated C5b-9 depositions induced by 20 µg/mL SARS-CoV-2 S1 (A) and S2 (C) were completely blocked in the presence of 1 µM factor D inhibitor (ACH145951) or 50 µg of anti-C5 antibody. C3c depositions induced by 20 µg/mL S1 (B) and S2 (D) were significantly reduced by factor D inhibitor but not by anti-C5 antibody. (E) A representative flow cytometry analysis demonstrated that 1 µM factor D inhibitor (dark blue curve) completely blocked the C5b-9 (left panel) and C3c deposition (right panel) triggered by 20 µg/mL S1 proteins (red curve). C5 inhibition with 50 µg of anti-C5 antibody (cyan curve) prevented S1-induced C5b-9 deposition, but not C3c accumulation. All experiments were repeated 3 times. Unpaired Student t test P values indicate statistical significance (*P < .05, **P < .01).
C5 and factor D inhibition block complement activation induced by SARS-CoV-2 spike proteins. Flow cytometry demonstrated C5b-9 depositions induced by 20 µg/mL SARS-CoV-2 S1 (A) and S2 (C) were completely blocked in the presence of 1 µM factor D inhibitor (ACH145951) or 50 µg of anti-C5 antibody. C3c depositions induced by 20 µg/mL S1 (B) and S2 (D) were significantly reduced by factor D inhibitor but not by anti-C5 antibody. (E) A representative flow cytometry analysis demonstrated that 1 µM factor D inhibitor (dark blue curve) completely blocked the C5b-9 (left panel) and C3c deposition (right panel) triggered by 20 µg/mL S1 proteins (red curve). C5 inhibition with 50 µg of anti-C5 antibody (cyan curve) prevented S1-induced C5b-9 deposition, but not C3c accumulation. All experiments were repeated 3 times. Unpaired Student t test P values indicate statistical significance (*P < .05, **P < .01).
SARS-CoV-2 S1 spike protein requires the cell surface to activate the APC
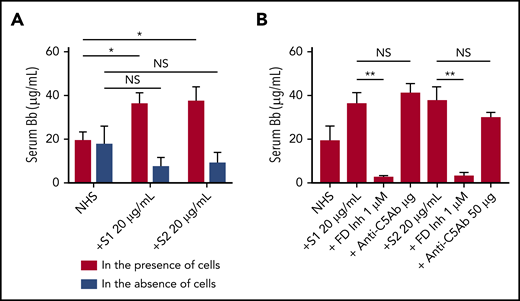
We next tested whether the activation of the APC induced by spike proteins was in fluid phase or on cell surfaces. Serum level of complement factor Bb, a specific marker of alternative pathway activation, was markedly elevated in the supernatant from TF1PIGAnull cells incubated with S1 and S2 (Figure 4A). The elevated Bb concentration was inhibited by ACH145951 but not anti-C5Ab (Figure 4B). The addition of S1 or S2 to human serum without TF1PIGAnull cells did not increase Bb levels, demonstrating that the spike proteins activate the APC predominantly on the cell surface and not in the fluid phase.
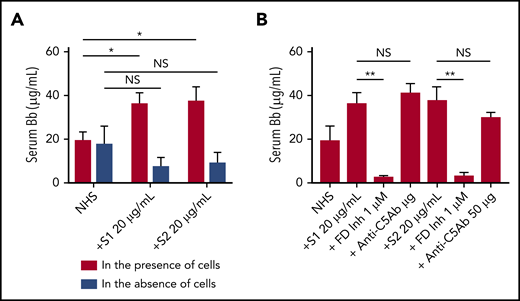
Serum level of factor Bb increases upon NHS activation by SARS-CoV-2 spike proteins in the presence of cells. Serum Bb concentration was measured by enzyme-linked immunosorbent assay (ELISA) after incubation of SARS-CoV-2 S1 and S2 with NHS in the presence and absence of TF1PIGAnull cells in APC-specific buffer. (A) S1 and S2 proteins significantly increased the Bb concentrations in serum when cells were present. Incubating NHS with S1 and S2 in the absence of cells did not significantly elevate serum Bb concentration from the baseline NHS level. (B) Increased Bb level in the cellular phase was completely blocked by 1 µM factor D inhibitor but not by anti-C5 antibody. All experiments were repeated 3 times. Unpaired Student t test P values indicate statistical significance (*P < .05, **P < .01, NS, not significant).
Serum level of factor Bb increases upon NHS activation by SARS-CoV-2 spike proteins in the presence of cells. Serum Bb concentration was measured by enzyme-linked immunosorbent assay (ELISA) after incubation of SARS-CoV-2 S1 and S2 with NHS in the presence and absence of TF1PIGAnull cells in APC-specific buffer. (A) S1 and S2 proteins significantly increased the Bb concentrations in serum when cells were present. Incubating NHS with S1 and S2 in the absence of cells did not significantly elevate serum Bb concentration from the baseline NHS level. (B) Increased Bb level in the cellular phase was completely blocked by 1 µM factor D inhibitor but not by anti-C5 antibody. All experiments were repeated 3 times. Unpaired Student t test P values indicate statistical significance (*P < .05, **P < .01, NS, not significant).
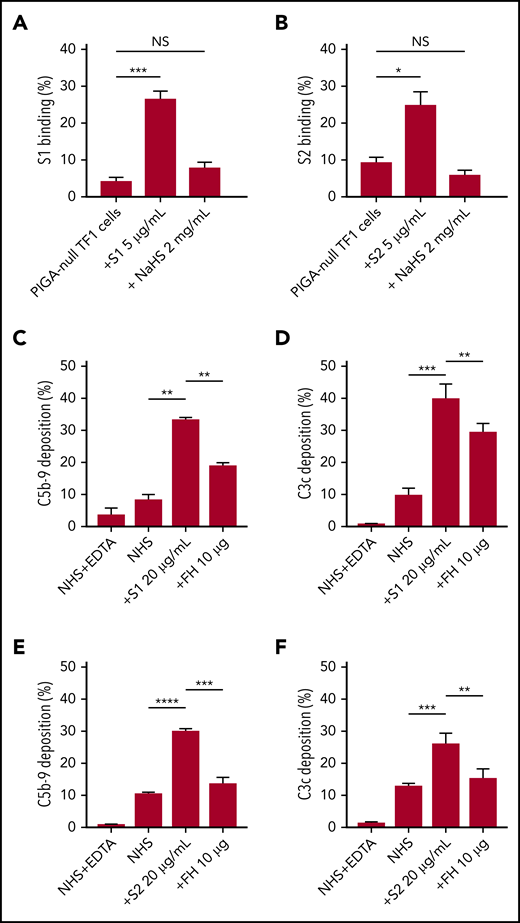
SARS-CoV-2 S1 and S2 spike protein bind to the HS on the cell surface
As an erythroblast cell line, TF1PIGAnull does not express angiotensin-converting enzyme-2, the entry receptor for SARS-CoV-2 (supplemental Figure 5), which makes it a good model for the study of spike protein–glycosaminoglycan interaction.29 Several groups have proved that the spike protein of SARS-CoV-2 binds to HS but not α2,3 or α2,6 sialic acids by microarray.30,31 Additional research showed that there are 3 HS-binding sites on the spike protein: 1 located inside the RBD of S1, 1 located in S2, and another 1 located at the S1/S2 cleavage site.32 Soluble HS has been shown to effectively block the binding of viruses that use HS on cell surfaces, therefore we hypothesized that this would also be true for SARS-CoV-2.33 Both S1 and S2, but not HCoV-OC43, bind TF1PIGAnull cells (supplemental Figure 3), however, S1 and S2 binding was significantly blocked by free HS (Figure 5A-B). Treatment with sialidase, which removes all α2-3–, α2-6–, and α2-8–linked sialic acid residues, did not result in any decrease in spike protein binding (supplemental Figure 4). These results confirm that HS is required for spike protein binding to cell surfaces and are consistent with recently published microarray data.30,31
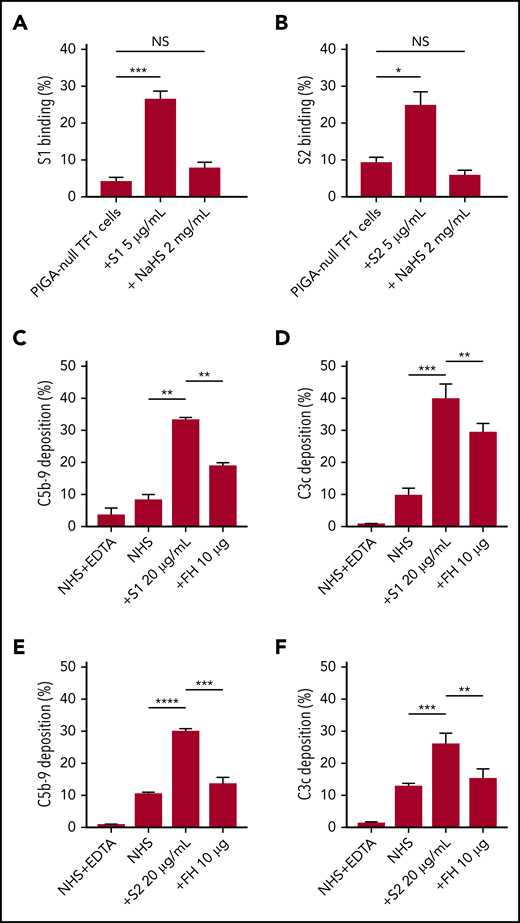
SARS-CoV-2 spike proteins bind to HS and interfere with factor H function. Coincubating 5 µg/mL His-tagged SARS-CoV-2 spike proteins with 2 mg/mL HS solution completely blocked the binding of S1 subunit (A) and S2 subunit (B) to TF1PIGAnull cells. Supplementing NHS with 10 µg of purified factor H protein significantly inhibited C5b-9 (C) and C3c deposition (D) triggered by 20 µg/mL S1 in APC-specific buffer. Addition of 10 µg of purified factor H also significantly inhibited the S2-induced C5b-9 deposition (E) and C3c deposition (F) in APC-specific buffer. All experiments were repeated at least 3 times. Unpaired Student t test P values indicate statistical significance (*P < .05, **P < .01, ***P < .001, ****P < .0001). FH, purified factor H protein; NaHS, HS sodium solution.
SARS-CoV-2 spike proteins bind to HS and interfere with factor H function. Coincubating 5 µg/mL His-tagged SARS-CoV-2 spike proteins with 2 mg/mL HS solution completely blocked the binding of S1 subunit (A) and S2 subunit (B) to TF1PIGAnull cells. Supplementing NHS with 10 µg of purified factor H protein significantly inhibited C5b-9 (C) and C3c deposition (D) triggered by 20 µg/mL S1 in APC-specific buffer. Addition of 10 µg of purified factor H also significantly inhibited the S2-induced C5b-9 deposition (E) and C3c deposition (F) in APC-specific buffer. All experiments were repeated at least 3 times. Unpaired Student t test P values indicate statistical significance (*P < .05, **P < .01, ***P < .001, ****P < .0001). FH, purified factor H protein; NaHS, HS sodium solution.
Factor H decreases C3c and C5b-9 deposition caused by S1 and S2 spike proteins
Apart from virus interaction, HS on cell surfaces is also important for binding factor H, a negative regulator of the APC.34,35 Thus, we hypothesized that S1 and S2 may be disrupting factor H binding or activity on the cell surface. We saturated the HS sites on cells with exogenous factor H proteinto overcome the APC activation by S1 and S2. As shown in our data, addition of purified factor H protein to NHS incubated with S1 or S2 decreased C5b-9 and C3c deposition on TF1PIGAnull cells (Figure 5C-F). Taken together, these data demonstrate that the S1 and S2 subunit binds to HS and appears to interfere with factor H function.
Discussion
COVID-19 is a life-threatening infectious disease that often results in hypercoagulability, thrombotic microangiopathy, and severe endothelial damage.12,36,37 The role of complement activation and its contribution to disease severity is increasingly recognized, but the mechanism of complement activation was unknown.38 Here, we demonstrate that SARS-CoV-2 spike protein, but not spike protein from a benign human coronavirus CoV-OC43 or the SARS-CoV-2 N protein, activates the APC on cell surfaces. Both S1 and S2 subunits activate the APC, but only in the presence of cells, demonstrating that APC activation is occurring on the cell surface and not the fluid phase. Structure resolution of the RBD in the SARS-CoV-2 spike protein S1 subunit shows a small positively charged region that may bind to negatively charged HS.39 Our findings confirm that both spike protein S1 and S2 subunits bind to HS, but counter previous speculation that SARS-CoV-2 N protein activates the lectin pathway.40 Although SARS-CoV-2 N protein shares some structural similarity with the N proteins from SARS-CoV and Middle Eastern respiratory syndrome–CoV, it may not necessarily have the same ability to bind mannan-binding lectin serine protease 2 and activate the lectin pathway. Soluble HS, but not sialidase treatment of target cells, blocked spike proteins from binding target cells.
ACH145951, a small molecule factor D inhibitor, was able to block the APC activation triggered by SARS-CoV-2 spike proteins (Figures 3 and 4). Once C3b attaches to a cell, it binds to and changes the conformation of factor B, which will then be cleaved by factor D to generate the APC C3 convertase (C3bBb)41 ; factor H can also bind to C3b and help factor I cleave C3b into inactivated C3b, thus factor D can be viewed as immediately upstream of factor H. Our data show that the factor D inhibitor (ACH145951) blocks cell-surface deposition of C3 fragments, C5b-9, and cell killing in the mHam (Figure 1 and 3). In addition, C5 antibody also blocks mHam killing and C5b-9 deposition but does not block deposition of C3 fragments or the release of Bb into serum, supporting complement activation primarily through the APC.
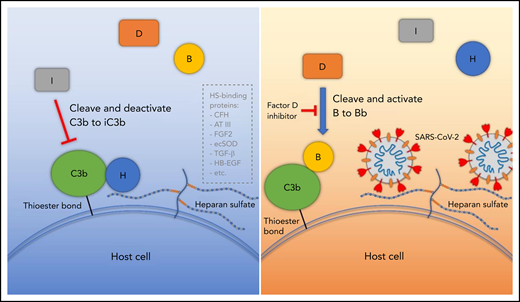
Factor H, a negative regulator of the APC, achieves a more active conformation when it binds to cells through interactions with glycosaminoglycans (HS and α2-3 N-linked sialic acid residues).35,42,43 Factor H is made up of 20 complement control protein (CCP) modules (also referred to as sushi domains or short consensus repeats). CCP 1-4 and CCP 19-20 bind to C3b, CCP 6-8 bind HS, and CCP 19-20 bind HS and sialic acids.44,45 Several human diseases, such as aHUS, age-related macular degeneration (AMD), HELLP syndrome, and CAPS, are associated with genetic variants that affect factor H function.20,46,47 Shiga toxin from enterohemorrhagic E coli also leads to endothelial damage and has been shown to bind to CCP 6-8 or CCP 18-20 of factor H and impair regulation of the APC on cell surfaces.48 Germline variants affecting CCP 19 and 20 in factor H predispose to aHUS and polymorphism in CCP 7 is found in up to 35% of patients with AMD.49,50 Interestingly, a history of AMD was recently reported to be a leading risk factor for intubation and death in COVID-19 patients.51 Our binding assays (Figure 5) suggest that SARS-CoV-2 spike proteins bind cell-surface HS, preventing factor H from achieving its optimal conformation to accelerate decay of the APC C3 convertase (Figure 6). Our rescue experiments also show that addition of purified factor H to serum decreased C3c and C5b-9 deposition on spike protein–treated cells, suggesting factor H as a key player in the APC activation by the spike protein. Whether spike protein directly blocks factor H from binding to HS or indirectly affects the conformation of factor H, however, remains to be further explored.
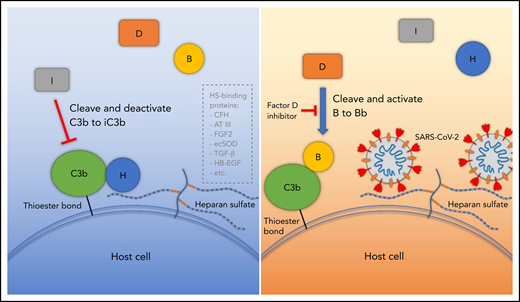
Proposed model for SARS-CoV-2 induced APC activation. Under normal conditions, factor H binds to HS on the cell surface and interacts with C3b, which facilitates factor I cleavage and deactivation of C3b. Upon infection, SARS-CoV-2 spike protein binds to HS on the cell surface and interferes with factor H function, which facilitates factor B binding to C3b and cleavage by factor D. AT III, antithrombin III; CFH, factor H; ecSOD, extracellular superoxide dismutase; FGF2, fibroblast growth factor 2; HB-EGF, heparin-binding epidermal growth factor; TGF-β, transforming growth factor β; VEGF, vascular endothelial growth factor.
Proposed model for SARS-CoV-2 induced APC activation. Under normal conditions, factor H binds to HS on the cell surface and interacts with C3b, which facilitates factor I cleavage and deactivation of C3b. Upon infection, SARS-CoV-2 spike protein binds to HS on the cell surface and interferes with factor H function, which facilitates factor B binding to C3b and cleavage by factor D. AT III, antithrombin III; CFH, factor H; ecSOD, extracellular superoxide dismutase; FGF2, fibroblast growth factor 2; HB-EGF, heparin-binding epidermal growth factor; TGF-β, transforming growth factor β; VEGF, vascular endothelial growth factor.
That SARS-CoV-2 spike proteins activate the APC has profound implications for understanding the multiorgan dysfunction, coagulopathy, and endothelial injury characteristic of COVID-19. Increased levels of C5a and soluble C5b-9 are detected in patients with moderate to severe COVID-19.52 Patients with COVID-19 also develop renal failure and some have biopsy-proven thrombotic microangiopathies.11,17 Thrombosis that is only partially responsive to anticoagulation (resistance to heparin treatment and thrombosis that develops despite appropriate prophylactic anticoagulation) is common in COVID-19 and characteristic of complementopathies,53,54 such as paroxysmal nocturnal hemoglobinuria,55 cold-agglutinin disease,56 and CAPS.20 In addition, HS is a binding partner for antithrombin III, which could further increase hypercoagulability in COVID-19 and may explain the heparin resistance that is frequently encountered in these patients (Figure 6).57 HS also interacts with many extracellular proteins, including fibroblast growth factor 2, vascular endothelial growth factor, transforming growth factor β, heparin-binding epidermal growth factor, and extracellular superoxide dismutase,58 suggesting a broad influence by the SARS-CoV-2 spike protein.
Our data may explain why only a minority of COVID-19 patients develop life-threatening multiorgan failure. Complement-driven diseases such as aHUS, HELLP, and CAPS often have underlying germline variants that impair the ability of endothelial cells to protect themselves from complement-mediated injury in the setting of a complement-amplifying condition such as pregnancy, cancer, autoimmunity, or other inflammatory states.28,59 A recent proteomics study by Shen et al with a machine learning model confirmed that complement proteins are actively involved in the acute phase of virus infection, including complement 6 and complement factor B, properdin, and carboxypeptidase N catalytic chain.60 Thus, it will be important to determine whether patients with the most severe forms of COVID-19 harbor variants in complement-regulatory genes. Already, a history of AMD, a disease associated with a common factor H variant, was identified as a major risk factor for mortality in COVID-19.51 Our data also have therapeutic implications for COVID-19 and raise the prospect for targeted therapy. Multinational randomized controlled trials of the C5 monoclonal antibody, ravulizumab (phase 3, NCT04369469), are already enrolling patients; however, our data demonstrate that C5 inhibition will prevent C5b-9 accumulation but not upstream complement activation induced by the SARS-CoV-2 spike proteins. Dense C3b deposition can lead to breakthrough from C5 monoclonal antibodies.61,62 Thus, complement inhibitors that bind upstream of factor H may be more specific and effective.27,63
The authors have not generated any genomic data in this study. Please contact brodsro@jhmi.edu for original data pertaining to this manuscript. No identifiable data will be shared. Requests for deidentified data from internal or external investigators will be evaluated on an individual basis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgment
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant R01 HL 133113 (R.A.B.).
Authorship
Contribution: J.Y., X.Y., and H.C. designed and performed experiments, interpreted data, and drafted the manuscript; S.C. and E.M.B. interpreted results and edited the manuscript; and R.A.B. designed the study, supervised the experiments, interpreted results, and wrote the manuscript.
Conflict-of-interest disclosure: R.A.B. has served on advisory board for Alexion Pharmaceutical Inc. The remaining authors declare no competing financial interests.
Correspondence: Robert A. Brodsky, Division of Hematology, School of Medicine, Johns Hopkins University, 720 Rutland Ave, Ross Research Building, Room 1025, Baltimore, MD 21205; e-mail: brodsro@jhmi.edu.