

In this issue of Blood, 1 provide novel molecular insights into the requirement for Runx1 in a mouse model of inv(16) acute myeloid leukemia (AML).
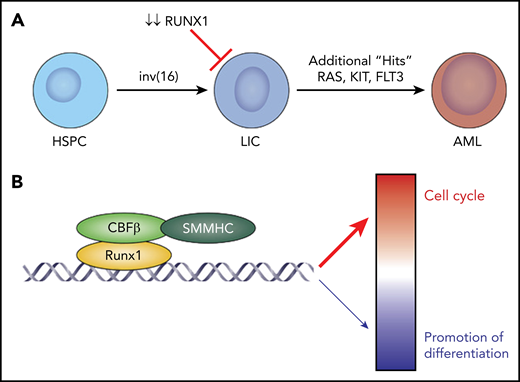
CBFβ-SMMHC requires RUNX1 for leukemia development. (A) Under normal conditions, hematopoietic stem and progenitor cells (HSPC) are converted to LICs by the action of the inv(16)-derived fusion oncoprotein, CBFβ-SMMHC, in combination with an activating mutation in a signaling molecule, such as NRAS, KRAS, or FLT3. The existing paradigm has been that CBFβ-SMMHC drives leukemia development by acting as a dominant negative which sequesters RUNX1, preventing it from promoting normal myeloid development. Zhen et al demonstrate that loss of RUNX1 prevents leukemia development in mice by preventing the development and maintenance of AMPs, a type of LIC specific to inv(16). (B) In AMP cells, RUNX1 recruits CBFβ-SMMHC to chromatin, where RUNX1:CBFβ-SMMHC predominantly activate cell cycle gene expression while simultaneously repressing a smaller number of genes that promote differentiation.
CBFβ-SMMHC requires RUNX1 for leukemia development. (A) Under normal conditions, hematopoietic stem and progenitor cells (HSPC) are converted to LICs by the action of the inv(16)-derived fusion oncoprotein, CBFβ-SMMHC, in combination with an activating mutation in a signaling molecule, such as NRAS, KRAS, or FLT3. The existing paradigm has been that CBFβ-SMMHC drives leukemia development by acting as a dominant negative which sequesters RUNX1, preventing it from promoting normal myeloid development. Zhen et al demonstrate that loss of RUNX1 prevents leukemia development in mice by preventing the development and maintenance of AMPs, a type of LIC specific to inv(16). (B) In AMP cells, RUNX1 recruits CBFβ-SMMHC to chromatin, where RUNX1:CBFβ-SMMHC predominantly activate cell cycle gene expression while simultaneously repressing a smaller number of genes that promote differentiation.
Core binding factor (CBF) leukemias are a common subtype of de novo AML and are represented by 2 distinct chromosomal abnormalities, t(8;21) and inv(16), which result in the fusion oncoproteins RUNX1-RUNX1T1 and CBFβ-SMMHC, respectively.2-5 They derive their name from normal CBF, which is a heterodimeric transcription factor that regulates gene expression and is composed of a DNA-binding α subunit (RUNX1, RUNX2, or RUNX3) and the non–DNA-binding CBFβ subunit. Although the 2 leukemias are treated similarly with high-dose chemotherapy and generally are considered favorable risk, up to 40% of patients relapse and subsequently require additional therapies, including allogeneic bone marrow transplantation, to achieve long-term survival. Relapses are due to the presence of a small number of leukemia-initiating cells (LICs; also called leukemia stem cells), which are resistant to conventional chemotherapy. Collectively, this illustrates the need for novel targeted therapies to improve survival while reducing toxicities for inv(16) AML patients.
Inv(16) has generally been considered a classic example of the original Gilliland 2-hit model of AML in which a proliferative signal is provided by a mutation, resulting in an activated signaling molecule, in combination with the loss of a transcription factor, which promotes the expansion of undifferentiated cells.6 In line with this model, CBFβ-SMMHC has been thought to cause AML by sequestering RUNX1, preventing it from regulating myeloid maturation and resulting in a differentiation block.3,7 This would suggest that a complete loss of RUNX1 activity is required for inv(16)-driven leukemia; however, there are other contradictory lines of evidence suggesting that RUNX1 may play a role in leukemogenesis. One example is that loss-of-function mutations in RUNX1 are often found in t(8;21), whereas they are rarely seen in inv(16), implying that there may be a negative selection bias against additional reduction in RUNX1 activity.4,5 As such, whether inv(16) requires some attenuated amount of RUNX1 to drive leukemia remains an important and unresolved question. This information is critical because it provides an important opportunity for developing targeted therapies.
Zhen et al used a genetically engineered “knock-in” allele that inducibly recapitulates inv(16) in mice (Mx1-Cre;Cbfb+/56M), in combination with an inducible null-allele of Runx1 (Runx1fl/fl), to definitively determine whether Runx1 is required for AML driven by CBFβ-SMMHC. Although the inv(16) animals with normal Runx1 developed AML with a latency of 16 weeks, similar to prior studies,8 animals deficient in Runx1 did not develop AML for up to 1 year, demonstrating that loss of Runx1 blocks CBFβ-SMMHC–driven leukemia development. Given the block in leukemia development, the investigators hypothesized that LICs were negatively affected by loss of Runx1. To test this hypothesis, they quantified a previously described LIC population termed abnormal myeloid progenitors (AMPs; Lin−c-Kit+Sca1−CD34−FCγII/III+).8 Importantly, the investigators found that loss of Runx1 hindered the development and maintenance of the AMP population, implying that Runx1 is required for CBFβ-SMMHC–driven LICs (see figure panel A).
Given the prior paradigm that CBFβ-SMMHC prevents RUNX1 from binding DNA, the investigators performed a series of detailed molecular studies to delineate the requirement for CBFβ-SMMHC and RUNX1 in the LIC population. Using bulk and single-cell RNA-sequencing, the investigators identified 2 hallmarks of LICs generated by CBFβ-SMMHC but lacking RUNX1. First, these LICs showed reduced expression of genes known to be critical drivers of the cell cycle, including the G2M checkpoint and targets of c-Myc and E2F. Second, they showed increased expression of genes required for normal myeloid differentiation. Collectively, these data imply that CBFβ-SMMHC works collaboratively with RUNX1 to mediate the proliferative signal and differentiation block, which are hallmarks of the Gilliland model for AML development.
Next, to definitively establish how CBFβ-SMMHC and RUNX1 cooperate to control gene expression, the investigators demonstrate that RUNX1 recruits CBFβ-SMMHC to chromatin, and they work in concert to activate gene expression, including genes that control proliferation (see figure panel B). This is in sharp contrast to prior literature that reported that CBFβ-SMMHC operates predominantly to repress gene expression. These important mechanistic studies connect how CBFβ-SMMHC and RUNX1 work collaboratively to directly regulate genes critical to LICs. It is important to note that these data represent 1 of the important ways that CBFβ-SMMHC operates during leukemia development, but it may be that the existing model of CBFβ-SMMHC sequestering RUNX1 occurs in parallel and is also critical to leukemogenesis. As such, it may be that, although CBFβ-SMMHC sequesters most RUNX1 during leukemia development, a small amount of active RUNX1 is required for transformation.
Perhaps most importantly, this work has the potential to inform targeted therapies. Although current treatment regimens rely on high-dose chemotherapy, an ongoing concern remains that conventional therapeutics are not effective at targeting LICs, which can cause relapse even when present in miniscule amounts. Prior studies have identified a small molecular inhibitor of the CBFβ-SMMHC:RUNX1 protein:protein interaction as a potential therapy for inv(16) AML.9,10 Zhen et al elevate this potential approach by demonstrating that it could directly target LICs, an important driver of relapse. In doing so, the investigators provide a path forward to develop more effective and less toxic therapies for patients with inv(16) AML.
Conflict-of-interest disclosure: The author declares no competing financial interests.

