

Key Points
Runx1 is required for leukemia-initiating cells in Cbfb-MYH11 mice.
RUNX1 recruits CBFβ-SMMHC to target genes.

Abstract
Inversion of chromosome 16 is a consistent finding in patients with acute myeloid leukemia subtype M4 with eosinophilia, which generates a CBFB-MYH11 fusion gene. It is generally considered that CBFβ-SMMHC, the fusion protein encoded by CBFB-MYH11, is a dominant negative repressor of RUNX1. However, recent findings challenge the RUNX1-repression model for CBFβ-SMMHC–mediated leukemogenesis. To definitively address the role of Runx1 in CBFB-MYH11–induced leukemia, we crossed conditional Runx1 knockout mice (Runx1f/f) with conditional Cbfb-MYH11 knockin mice (Cbfb+/56M). On Mx1-Cre activation in hematopoietic cells induced by poly (I:C) injection, all Mx1-CreCbfb+/56M mice developed leukemia in 5 months, whereas no leukemia developed in Runx1f/fMx1-CreCbfb+/56M mice, and this effect was cell autonomous. Importantly, the abnormal myeloid progenitors (AMPs), a leukemia-initiating cell population induced by Cbfb-MYH11 in the bone marrow, decreased and disappeared in Runx1f/fMx1-CreCbfb+/56M mice. RNA-seq analysis of AMP cells showed that genes associated with proliferation, differentiation blockage, and leukemia initiation were differentially expressed between Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice. In addition, with the chromatin immunocleavage sequencing assay, we observed a significant enrichment of RUNX1/CBFβ-SMMHC target genes in Runx1f/fMx1-CreCbfb+/56M cells, especially among downregulated genes, suggesting that RUNX1 and CBFβ-SMMHC mainly function together as activators of gene expression through direct target gene binding. These data indicate that Runx1 is indispensable for Cbfb-MYH11–induced leukemogenesis by working together with CBFβ-SMMHC to regulate critical genes associated with the generation of a functional AMP population.
Introduction
Inversion of chromosome 16, inv(16), is associated with acute myeloid leukemia (AML) subtype M4 with eosinophilia, which generates the CBFB-MYH11 fusion gene.1 CBFβ-SMMHC, encoded by CBFB-MYH11, comprises most of core-binding factor β (CBFβ, 1-165 amino acids of a total of 187 amino acids) and the C-terminal coiled-coil domain of the smooth muscle myosin heavy chain (SMMHC).2,3 Studies have shown that CBFβ-SMMHC is necessary, but not sufficient for leukemogenesis.4,5
CBFβ-SMMHC is thought to initiate leukemogenesis by blocking normal hematopoietic differentiation through inhibiting the function of RUNX1, which is 1 of the 3 α-subunits in the CBF family (RUNX1, RUNX2, and RUNX3), all of which have a RUNT domain that mediates DNA binding and heterodimerization with CBFβ.6 Binding with CBFβ leads to the stabilization of the RUNX–DNA interaction and gene expression regulation through the transactivation domain of RUNX proteins.7 CBFβ-SMMHC retains the RUNX-binding domain from CBFβ and also contains a second RUNX high-affinity binding domain in the SMMHC tail, resulting in a higher binding affinity for RUNX1 than wild-type CBFβ.8 In vitro studies have shown that CBFβ-SMMHC may serve as a transcriptional repressor by sequestering RUNX1 in the cytoplasm.9-11 In vivo, Cbfb-MYH11 heterozygous knockin mice (Cbfb+/MYH11) have a complete block in definitive hematopoiesis and central nervous system hemorrhaging at embryonic day 12.5 (E12.5), which contributes to lethality by E13.5.12 This phenotype is similar to that of Runx1 null (Runx1−/−) and Cbfb null (Cbfb−/−) mice,13-17 suggesting that CBFβ-SMMHC acts as a dominant repressor of RUNX1 and CBFβ functions during embryogenesis. However, subsequent studies challenge the RUNX1 repression model for CBFβ-SMMHC–mediated leukemogenesis, including (1) loss-of-function mutations in RUNX1 are common in human AML, but they have not been found in inv(16) AML18-21 ; (2) Cbfb+/MYH11 embryos have differentiation defects and abnormal gene expression during primitive hematopoiesis that are not see in Runx1−/−or Cbfb−/− embryos22 ; (3) there are both upregulated and downregulated genes in CBFβ-SMMHC–expressing cells23 ; (4) many genes are uniquely deregulated in Cbfb+/MYH11 embryos but not in Runx1−/−or Cbfb−/− embryos and are expressed in leukemic cells from mice and humans22 ; (5) knock-in mice expressing a high-affinity binding domain–deleted CBFβ-SMMHC developed leukemia faster than mice expressing full-length CBFβ-SMMHC, although hematopoietic defects associated with Runx1 inhibition were partially rescued24 ; and (6) in the Cbfb-MYH11 expressing ME-1 cells, 89% of CBFβ-SMMHC binding regions colocalize with RUNX1 binding sites.25
More importantly, our previous work demonstrated that RUNX1 activity is required for Cbfb-MYH11–induced differentiation defects during both primitive and definitive hematopoiesis, and insufficient Runx1 activity (Runx1+/lz) delays Cbfb-MYH11–induced leukemia development in mice.26 Together, these results suggested that CBFβ-SMMHC does not simply repress RUNX1. Instead, current data imply that Runx1 is required for CBFB-MYH11 leukemogenesis.
However, this previous work was inconclusive because the Runx1+/lz mice used in the previous study contained an artificial Runx1-lz allele and 1 wild-type Runx1 allele, which still retains some Runx1 function.26
To definitely address the functional importance of Runx1 in Cbfb-MYH11–induced leukemia, we used a Cre-based conditional Runx1 knockout mice (Runx1f/f)27 and generated Cbfb-MYH11 knockin mice with homozygous Runx1 deletion. We found that the resulting Runx1−/−Cbfb+/MYH11 mice could not develop leukemia, and the likely reason was dysregulation of critical genes in the abnormal myeloid progenitor (AMP) population, including those associated with proliferation, differentiation blockage, and maintenance of leukemia-initiating ability of such cells. In addition, our results suggested that, besides acting as a dominant negative repressor of RUNX1, CBFβ-SMMHC also acts as a coactivator of RUNX1 target genes. These results indicate that Runx1 is indispensable for leukemia development induced by Cbfb-MYH11, and the RUNX1–CBFβ-SMMHC interaction could be a good target for developing novel treatments of inv(16) leukemia.
Methods
Animals
All animals used in this study were approved by the National Human Genome Research Institute Animal Care and Use Committee, and all the procedures performed followed relevant National Institutes of Health guidelines and regulations. Cbfb-MYH11 conditional knockin (Cbfb+/56M),28 Mx1-Cre mice,29 and Runx1 conditional knockout (Runx1f/f) mice27 have been described previously. All mice were genotyped by polymerase chain reaction (PCR) with gene-specific primers (supplemental Table 1 available on the Blood Web site) using tail-snip DNA prepared with DNeasy Blood & Tissue Kit (Qiagen). Eight- to 12-week-old mice and their littermate controls were injected intraperitoneally with 250 μg poly (I:C) (pIpC; InvivoGen) to induce the expression of Cbfb-MYH11 and/or knockout of Runx1 every other day for 3 doses. For the noncompetitive transplantation assay, C57BL/6 (CD45.1) × 129/SvEv F1 mice were irradiated (550 rads × 2 times) and then injected through tail vein with 1 million bone marrow cells from indicated donor mice. The recipient mice were then treated with pIpC as above 9 weeks after transplantation to induce the expression of Cbfb-MYH11 and deficiency of Runx1. All mice were observed for leukemia development for 12 months after pIpC injections.
Plasmids
RUNX1 cDNA sequence was cloned into pmCherry-C1 and pcDNA3.1(+) vector (Addgene), respectively. CBFB and CBFB-MYH11 cDNA sequences were cloned into the PGEM vector (Promega) and pEGFP-C1 vector (Addgene), respectively. Truncated RUNX1–related plasmids were created on related RUNX1 plasmids using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent).
Flow cytometry
Peripheral blood cells and spleen and bone marrow cells from mice were harvested and stained as described previously.23 See the supplemental Data (available on the Blood Web site) for detail for the antibodies used in this study.
Western blot analysis
Western blot analysis was performed with standard protocols. More details are described in the supplemental Methods.
Immunofluorescence
Immunofluorescence staining of transfected 293T cells and bone marrow cells from mice was performed with standard protocols. More details are described in the supplemental Methods.
CSF1R promoter reporter assay
The CSF1R promoter reporter assay was performed as described previously.30
Quantitative PCR
Quantitative PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) according to manufacturer’s instruction. Runx1 unexcised primers were used to detect unexcised Runx1 flox allele, and exon 5/6 primers were used to detect unexcised Cbfb-MYH11 flox allele. Genomic control primers were used as internal controls for genomic DNA. Primers are listed in supplemental Table 1.
RNA sequencing, ChIC sequencing, and single-cell RNA sequencing
Mice were injected with pIpC, and 2 to 3 weeks later, the AMP population in the bone marrow cells of these mice were sorted out with a BD FACSAria IIIu cell sorter (BD Biosciences). mRNA from the AMP cells was extracted and subjected to RNA sequencing (RNA-Seq). Paraformaldehyde (1%)-fixed AMP cells were subjected to chromatin immunocleavage sequencing (ChIC-seq) as described31 ; the 10X genomics chromium platform32 was used to capture isolated AMP cells, and all steps of single-cell RNA sequencing were performed according to manufacturer’s instructions. Detailed procedures and data analysis are provided in supplemental Methods.
Statistical analysis
Data were analyzed using Graphpad Prism. Results are expressed as mean ± standard error of the mean (SEM). Differences between the 2 groups were tested with the Student t test. The differences in survival times of mice were analyzed with the Kaplan-Meier method and log-rank test. P < .05 was considered statistically significant.
Results
Truncated RUNX1 protein in the Runx1f/f mice was functionally defective at the molecular and cellular levels
To determine whether RUNX1 is required during Cbfb-MYH11 leukemogenesis, we crossed a Cre-based conditional Runx1 knockout mouse strain (Runx1f/f)27 with Mx1-Cre–based conditional Cbfb-MYH11 knockin mice (Mx1-CreCbfb+/56M)28 to generate Runx1f/fMx1-CreCbfb+/56M mice. The conditional Runx1 knockout allele deletes Runx1 exon 4 (amino acids 158-192 of mouse RUNX1, equivalent to amino acids 171-205 of human RUNX1; supplemental Figure 1) after Cre-mediated recombination; however, a truncated RUNX1 protein could be detected in the bone marrow cells (supplemental Figure 2). Although it had been shown that a C-terminal truncation of human RUNX1 to amino acid 170 was sufficient to disrupt DNA and CBFβ binding in vitro,33 whether this internally truncated RUNX1 protein was functional remained to be determined.
As the nuclear localization sequence in RUNX1 was partially disrupted by the exon 4 deletion (supplemental Figure 1A),34 the localization of the truncated RUNX1 was examined. Indeed, unlike RUNX1 in wild-type and Mx1-CreCbfb+/56M mice, which was mainly present in the nuclear fraction, the truncated RUNX1 was found in both cytoplasmic and nuclear fractions in pIpC-treated Runx1f/fMx1-Cre and Runx1f/fMx1-CreCbfb+/56M bone marrow cells (Figure 1A; supplemental Figure 2). Similarly, in transfected 293T cells the truncated RUNX1 was located in both cytoplasm and nuclei (Figure 1B).
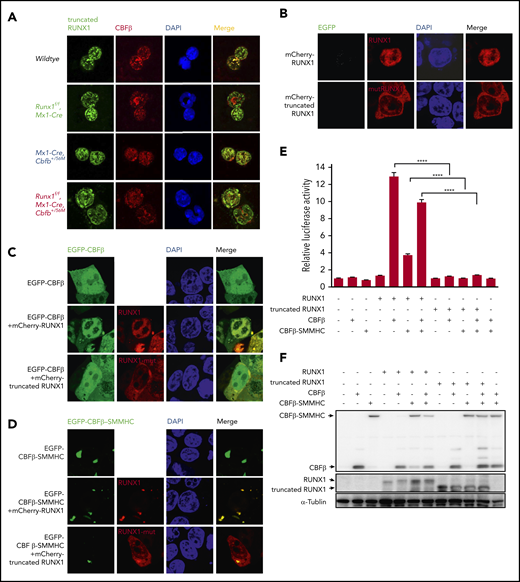
The mutated RUNX1 protein in Runx1f/fmice was nonfunctional. (A) Immunofluorescent staining of bone marrow cells from the indicated mice 2 weeks after pIpC treatment to detect the colocalization between RUNX1 or the mutated RUNX1 and CBFβ/CBFβ-SMMHC. (B-D) 293T cells were transfected with the indicated plasmids, and immunofluorescence staining was performed to detect the localization of the indicated proteins. The labels on the left side of the pictures indicate the plasmids transfected into the cells and the labels on top of the pictures indicate the observed proteins at the appropriate microscope filter settings. (E-F) Luciferase reporter assay in 293T cells transfected with a CSF1R promoter-driven luciferase reporter plasmid and plasmids encoding the indicated proteins. (E) Relative activities (mean ± SEM) were determined based on 3 independent experiments. ****P < .0001. (F) Representative expression levels of the transfected proteins for this reporter assay by western blot analysis.
The mutated RUNX1 protein in Runx1f/fmice was nonfunctional. (A) Immunofluorescent staining of bone marrow cells from the indicated mice 2 weeks after pIpC treatment to detect the colocalization between RUNX1 or the mutated RUNX1 and CBFβ/CBFβ-SMMHC. (B-D) 293T cells were transfected with the indicated plasmids, and immunofluorescence staining was performed to detect the localization of the indicated proteins. The labels on the left side of the pictures indicate the plasmids transfected into the cells and the labels on top of the pictures indicate the observed proteins at the appropriate microscope filter settings. (E-F) Luciferase reporter assay in 293T cells transfected with a CSF1R promoter-driven luciferase reporter plasmid and plasmids encoding the indicated proteins. (E) Relative activities (mean ± SEM) were determined based on 3 independent experiments. ****P < .0001. (F) Representative expression levels of the transfected proteins for this reporter assay by western blot analysis.
We then determined whether the truncated RUNX1 interacted with CBFβ and CBFβ-SMMHC. As expected, when full-length RUNX1 was cotransfected with CBFβ, the localization of CBFβ was changed compared with cells transfected with CBFβ alone, from both cytoplasm and nuclear localization to mainly nuclear, suggesting interaction between RUNX1 and CBFβ (Figure 1C). In contrast, when the truncated RUNX1 was cotransfected with CBFβ, the localization of CBFβ did not change compared with cells transfected with CBFβ alone, suggesting the truncated RUNX1 could not interact with CBFβ (Figure 1C). Similar subcellular localization changes were observed when CBFβ-SMMHC was cotransfected with truncated RUNX1 (Figure 1D). The lack of interaction between the truncated RUNX1 and CBFβ/CBFβ-SMMHC was also confirmed with endogenous proteins. As shown in Figure 1A, in Mx1-Cre, Cbfb+/56M bone marrow cells, clear colocalization signals were observed between RUNX1 and CBFβ/CBFβ-SMMHC in the nuclei, whereas in Runx1f/fMx1-Cre and Runx1f/fMx1-Cre Cbfb+/56M bone marrow cells, the truncated RUNX1 was located in both cytoplasm and nuclei, and much less colocalization signal was observed between the truncated RUNX1 and CBFβ/CBFβ-SMMHC. All together, these results suggested that the truncated RUNX1 lost the ability to interact with CBFβ and CBFβ-SMMHC.
We also tested the ability of the truncated RUNX1 to transactivate target genes in the presence of CBFβ or CBFβ-SMMHC in a reporter assay, in which the expression of luciferase was driven by the promoter of CSF1R.35 As can be seen in Figure 1E-F, the truncated RUNX1 was not able to transactivate CSF1R luciferase activity as RUNX1 did, whether in the absence or presence of the CBFβ proteins. Taken together, these results confirm that the truncated RUNX1 is a loss of function mutant.
Runx1 was required for Cbfb-MYH11–induced leukemia
To explore the effect of Runx1 knockout on Cbfb-MYH11–induced leukemia, we treated adult (8-12 weeks old) Mx1-CreCbfb+/56M (n = 9), Runx1f/fMx1-CreCbfb+/56M (n = 11), and Runx1f/fMx1-Cre (n = 6) mice, as well as their littermate control mice (wild type; Mx1-Cre or other genotypes without Mx1-Cre, n = 7) with pIpC to induce Mx1-Cre expression. The excisions of Runx1 and Cbfb-MYH11 flox alleles in bone marrow cells of these mice 4 months after pIpC treatment were nearly complete, as determined by quantitative PCR (supplemental Figure 3A).
As expected, Mx1-CreCbfb+/56M mice succumbed to AML with a median survival of 114 days (Figure 2A-D; supplemental Figure 4). Of the 11 Runx1f/fMx1-CreCbfb+/56M mice, 3 of them died around 2 weeks after pIpC treatment. The exact cause of death was not clear but all 3 developed pancytopenia, especially severe anemia and thrombocytopenia (supplemental Figure 5A), with no signs of leukemia (supplemental Figure 5B-C). In addition, transplantation of spleen cells from these mice did not lead to leukemia development in the recipient mice (supplemental Figure 5D).
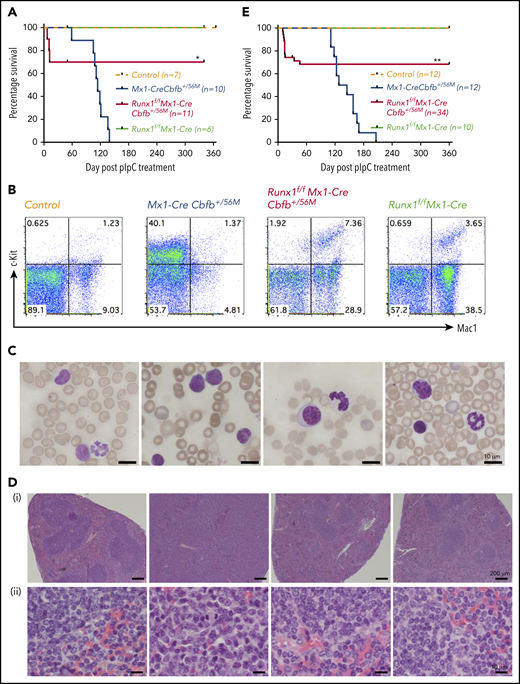
Runx1 is required for Cbfb-MYH11–induced leukemia. (A) Mice of the indicated genotype were treated with pIpC to induce the expression of Cbfb-MYH11 and/or Runx1 deficiency, and leukemia development in these mice was monitored for 1 year. Kaplan-Meier survival curves of these mice are shown. (B-D) Mice of the indicated genotypes were treated with pIpC and then killed 4 months after the last dose of pIpC for analysis. (B) Representative fluorescence-activated cell sorting (FACS) plots of c-Kit+ and Mac1+ cells in the peripheral blood of these mice. (C) Representative Wright-Giemsa–stained peripheral blood smears from these mice. (D) Representative hematoxylin and eosin–stained spleen sections (i, 50×; ii, 400×) from these mice. (E) Kaplan-Meier survival curves of recipient mice (3-5 per donor mouse) after noncompetitive transplantation assay. *P < .05 and **P < .01, each comparing the Runx1f/fMx1-CreCbfb+/56M group with the Mx1-CreCbfb+/56M group.
Runx1 is required for Cbfb-MYH11–induced leukemia. (A) Mice of the indicated genotype were treated with pIpC to induce the expression of Cbfb-MYH11 and/or Runx1 deficiency, and leukemia development in these mice was monitored for 1 year. Kaplan-Meier survival curves of these mice are shown. (B-D) Mice of the indicated genotypes were treated with pIpC and then killed 4 months after the last dose of pIpC for analysis. (B) Representative fluorescence-activated cell sorting (FACS) plots of c-Kit+ and Mac1+ cells in the peripheral blood of these mice. (C) Representative Wright-Giemsa–stained peripheral blood smears from these mice. (D) Representative hematoxylin and eosin–stained spleen sections (i, 50×; ii, 400×) from these mice. (E) Kaplan-Meier survival curves of recipient mice (3-5 per donor mouse) after noncompetitive transplantation assay. *P < .05 and **P < .01, each comparing the Runx1f/fMx1-CreCbfb+/56M group with the Mx1-CreCbfb+/56M group.
The remaining 8 Runx1f/fMx1-CreCbfb+/56M mice were free of hematopoietic malignancy 12 months after pIpC injection, with nearly complete excisions of Runx1 and Cbfb-MYH11 flox alleles in bone marrow cells at 12 months after pIpC injection (supplemental Figure 3B). This phenotype was different from that observed in Runx1+/lzMx1-CreCbfb+/56M mice, ∼80% of which developed leukemia, albeit with longer latency than Mx1-CreCbfb+/56M mice.26 As expected, no leukemia was observed in the Runx1f/fMx1-Cre mice or the control mice (Figure 2A). The Runx1f/fMx1-CreCbfb+/56M mice did develop nonlethal thrombocytopenia (supplemental Figure 4A) and mild myeloproliferative changes (Figure 2B,D; supplemental Figure 4B-C), which were likely caused by Runx1 knockout,27,36 as similar phenotypes were also observed in Runx1f/fMx1-Cre mice (Figure 2B,D; supplemental Figure 4). These results suggest that Runx1 is required for Cbfb-MYH11–induced leukemogenesis.
The requirement of Runx1 for Cbfb-MYH11 leukemogenesis was cell autonomous
To determine whether Runx1 is required in the hematopoietic cells or in the surrounding microenvironment for Cbfb-MYH11 leukemogenesis, we transplanted bone marrow cells from Mx1-CreCbfb+/56M, Runx1f/fMx1-CreCbfb+/56M, and Runx1f/fMx1-Cre mice into lethally irradiated recipient mice, which were injected with pIpC 9 weeks later to induce the expression of Cbfb-MYH11 and knockout of Runx1. Efficient engraftment of donor cells was confirmed in all recipient mice before pIpC treatment (supplemental Figure 6A). Mice transplanted with cells from Mx1-CreCbfb+/56M mice died of leukemia with a median survival of 136.5 days (Figure 2E; supplemental Figure S6B-C). In contrast, 30% of mice transplanted with cells from Runx1f/fMx1-CreCbfb+/56M mice died shortly after pIpC treatment (Figure 2E) with pancytopenia, including severe anemia and thrombocytopenia (supplemental Figure 6B-C), whereas the remaining 70% of the mice transplanted with Runx1f/fMx1-CreCbfb+/56M cells were free of leukemia up to 1 year after pIpC injection (Figure 2E), with nearly complete excisions of Runx1 and Cbfb-MYH11 flox alleles in bone marrow cells at 12 months after pIpC injection (supplemental Figure 6D). These results suggest that the requirement of Runx1 for Cbfb-MYH11 leukemogenesis is cell autonomous.
AMP cells could not initiate leukemia in the Runx1f/fMx1-CreCbfb+/56M mice
We then explored the potential mechanism of the Runx1 requirement for Cbfb-MYH11–induced leukemia development. As we showed previously, Cbfb-MYH11 expression in adult mice leads to aberrant hematopoiesis before leukemic transformation and these abnormalities include increased lineage negative (Lin−) cells and decreased myeloid progenitor cells (LK, Lin−/c-Kit+/Sca1−) in the bone marrow.22,28 These changes were partially rescued by Runx1 knockout (Figure 3A-B). Interestingly, we found that the AMP population (LK/CD34−/FcγRII/III+), which was absent in control and Runx1f/fMx1-Cre mice (Figure 3A; supplemental Figure 7A), was larger in the Runx1f/fMx1-CreCbfb+/56M mice compared with that in Mx1-CreCbfb+/56M mice shortly after pIpC treatment (2-3 weeks). However, the AMP population decreased 4 to 5 weeks after pIpC treatment and disappeared after 4 months (Figure 3C-D). On the other hand, the AMP population, but not common myeloid progenitors (CMP, LK/CD34+/FcγRII/III+), granulocyte-macrophage progenitor (GMP, LK/CD34+/FcγRII/IIIhigh), and megakaryocyte-erythroid progenitor (MEP, LK/CD34−/FcγRII/III−; Figure S7B), grew larger with time in the Mx1-CreCbfb+/56M mice, and eventually led to leukemia (Figure 3C-D). These results suggested that Runx1 was required for the generation and maintenance of a functional AMP population and the decrease/disappearance of this AMP population was likely responsible for the failure of leukemia development in the Runx1f/fMx1-CreCbfb+/56M mice.
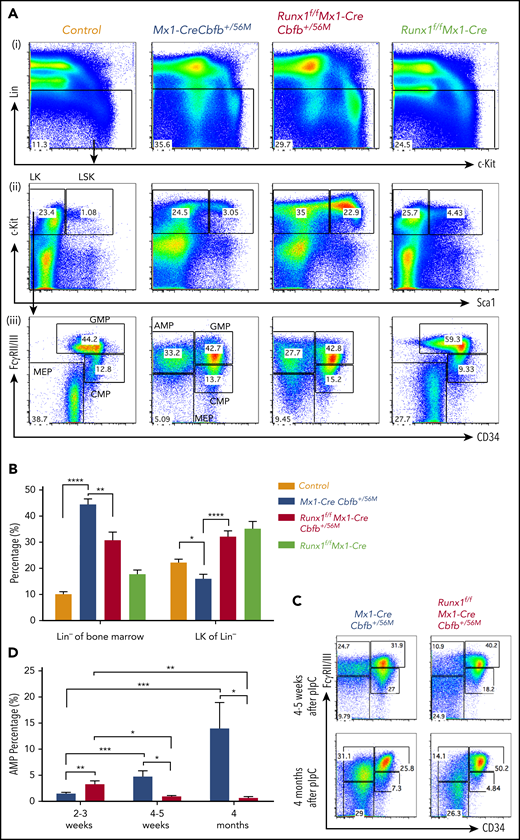
Runx1 is required for the maintenance of the AMP population. (A-D) The indicated groups of mice were treated with pIpC to induce the expression of Cbfb-MYH11 and/or Runx1 deficiency. At certain time points after pIpC treatment, the mice were killed, and flow cytometry assays were performed. (A) Representative FACS plots of bone marrow cells from mice treated with pIpC for 2 to 3 weeks gated on single cells (i), Lin− cells (ii), and LK cells (iii) are shown for the intensities of the indicated antibodies. (B) Bar graphs showing the percentages (mean ± SEM) of Lin− of bone marrow and LK fraction of Lin− compartments in mice of the indicated genotypes as showed in panel A. (C) Representative FACS plots of bone marrow cells gated on LK cells from mice treated with pIpC for 4 to 5 weeks and 4 months. (D) Bar graph showing the percentages (mean ± SEM) of the AMP population in the bone marrow of mice of indicated genotypes treated with pIpC for 2 to 3 weeks, 4 to 5 weeks, and 4 months as showed in panels A and C. *P < .05, **P < .01, ***P < .001, ****P < .0001. All genotypes in this figure are color-coded the same way (blue for Mx1-CreCbfb+/56M, red for Runx1f/fMx1-CreCbfb+/56M, and green for Runx1f/fMx1-Cre).
Runx1 is required for the maintenance of the AMP population. (A-D) The indicated groups of mice were treated with pIpC to induce the expression of Cbfb-MYH11 and/or Runx1 deficiency. At certain time points after pIpC treatment, the mice were killed, and flow cytometry assays were performed. (A) Representative FACS plots of bone marrow cells from mice treated with pIpC for 2 to 3 weeks gated on single cells (i), Lin− cells (ii), and LK cells (iii) are shown for the intensities of the indicated antibodies. (B) Bar graphs showing the percentages (mean ± SEM) of Lin− of bone marrow and LK fraction of Lin− compartments in mice of the indicated genotypes as showed in panel A. (C) Representative FACS plots of bone marrow cells gated on LK cells from mice treated with pIpC for 4 to 5 weeks and 4 months. (D) Bar graph showing the percentages (mean ± SEM) of the AMP population in the bone marrow of mice of indicated genotypes treated with pIpC for 2 to 3 weeks, 4 to 5 weeks, and 4 months as showed in panels A and C. *P < .05, **P < .01, ***P < .001, ****P < .0001. All genotypes in this figure are color-coded the same way (blue for Mx1-CreCbfb+/56M, red for Runx1f/fMx1-CreCbfb+/56M, and green for Runx1f/fMx1-Cre).
Gene expression changes in the AMP population in Runx1f/fMx1-CreCbfb+/56M mice
To understand how the AMP population disappeared in Runx1f/fMx1-CreCbfb+/56M mice, we performed RNA-seq with AMP cells isolated from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice (2-3 weeks after pIpC treatment) to profile gene expression changes between them. Principal component analysis showed a clear separation of the AMP cells in Mx1-CreCbfb+/56M mice from those in the Runx1f/fMx1-CreCbfb+/56M mice (Figure 4A). Compared with Mx1-CreCbfb+/56M mice, 858 genes were identified as differentially expressed genes (DEGs; q value < 0.01, absolute fold change ≥ 2) in Runx1f/fMx1-CreCbfb+/56M mice, with 69.7% upregulated genes and 30.3% downregulated genes (Figure 4B; supplemental Table 2).
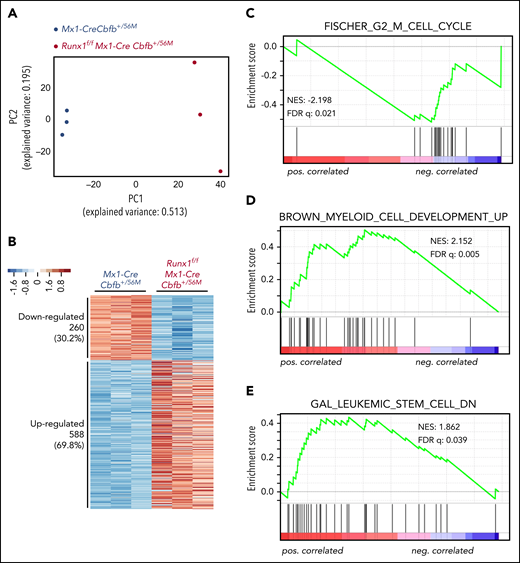
Runx1 is required for the regulation of critical genes to maintain functional AMPs for leukemogenesis by Cbfb-MYH11. (A-G) RNA-seq was performed on AMP cells isolated from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice 2 to 3 weeks after pIpC treatment. N = 3 for each genotype. (A) Two-dimensional principal component analysis plots showing clear separation between these 2 genotype groups. (B) Heatmap representation of identified DEGs between these 2 groups. Numbers and percentages of DEGs in each of the 2 expression clusters (down- and upregulated) are listed on the left. (C-E) GSEA (Preranked) identified curated genes sets significantly enriched in these DEGs, including (C) the FISCHER_G2_M_CELL_CYCLE gene set, which is negatively correlated with DEGs upregulated in Runx1f/fMx1-CreCbfb+/56M cells; (D) the BROWN_MYELOID_CELL_DEVELOPMENT_UP gene set, which is positively correlated with DEGs upregulated in Runx1f/fMx1-CreCbfb+/56M cells; and (E) the GAL_LEUKEMIC_STEM_CELL_DN gene set, which is positively correlated with DEGs upregulated in Runx1f/fMx1-CreCbfb+/56M cells.
Runx1 is required for the regulation of critical genes to maintain functional AMPs for leukemogenesis by Cbfb-MYH11. (A-G) RNA-seq was performed on AMP cells isolated from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice 2 to 3 weeks after pIpC treatment. N = 3 for each genotype. (A) Two-dimensional principal component analysis plots showing clear separation between these 2 genotype groups. (B) Heatmap representation of identified DEGs between these 2 groups. Numbers and percentages of DEGs in each of the 2 expression clusters (down- and upregulated) are listed on the left. (C-E) GSEA (Preranked) identified curated genes sets significantly enriched in these DEGs, including (C) the FISCHER_G2_M_CELL_CYCLE gene set, which is negatively correlated with DEGs upregulated in Runx1f/fMx1-CreCbfb+/56M cells; (D) the BROWN_MYELOID_CELL_DEVELOPMENT_UP gene set, which is positively correlated with DEGs upregulated in Runx1f/fMx1-CreCbfb+/56M cells; and (E) the GAL_LEUKEMIC_STEM_CELL_DN gene set, which is positively correlated with DEGs upregulated in Runx1f/fMx1-CreCbfb+/56M cells.
Gene set enrichment analysis (GSEA)37 was performed with the entire RNA-seq data of the AMP cells isolated from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice. As shown in supplemental Table 3, only 6 gene sets were enriched (with FDR < 0.25) in the AMP cells in the Mx1-CreCbfb+/56M mice. Interestingly, genes involved in regulating cell cycle were significantly enriched in these gene sets, including G2M checkpoint genes, MYC targets, and E2F targets. On the other hand, these cell cycle–related gene sets were not enriched in the AMP cells from the Runx1f/fMx1-CreCbfb+/56M mice. In fact, no gene sets were identified in the Runx1f/fMx1-CreCbfb+/56M AMP cells with FDR < 0.25. These findings may suggest that RUNX1 is a major driver of cell cycle in the AMP cells.
We also performed GSEA analysis of the DEGs between AMP cells isolated from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice. The data showed that several curated gene sets were significantly enriched. As shown in Figure 4C-D and supplemental Table 4, the upregulated DEGs in Runx1f/fMx1-CreCbfb+/56M cells were negatively correlated with the FISCHER_G2_M_CELL_CYCLE gene set (cell cycle genes with peak expression in G2/M check point, such as CDK1) and positively correlated with the BROWN_MYELOID_CELL_DEVELOPMENT_UP gene set (genes with potential to regulate myeloid cell growth and differentiation), suggesting that the disappearance of the AMP population in Runx1f/fMx1-CreCbfb+/56M mice was at least in part through inhibition of proliferation and promotion of differentiation. Moreover, the upregulated DEGs in Runx1f/fMx1-CreCbfb+/56M cells were positively correlated with the GAL_LEUKEMIC_STEM_CELL_DN gene set (Figure 4E; supplemental Table 4), which contained DEGs downregulated in leukemic stem cells, suggesting that the AMP population in Runx1f/fMx1-CreCbfb+/56M mice lost the leukemia initiating ability. In addition, gene ontology analysis showed that downregulated DEGs were associated with terms such as cell differentiation and mitotic cell cycle (supplemental Figure 8A), whereas the upregulated DEGs were associated with immune response (supplemental Figure 8B), consistent with the finding that RUNX1 can negatively regulate inflammatory cytokine production.38
IPA upstream analysis showed that GATA2 was the most significant putative upstream transcription factor that regulated the DEGs in the AMP cells of the Runx1f/fMx1-CreCbfb+/56M mice (supplemental Figure 8C), suggesting that RUNX1 functions in part through other key hematopoietic transcription factors, such as GATA2, consistent with our previous observation that Gata2 deficiency delays leukemogenesis by Cbfb-MYH11.39
RUNX1 and CBFβ-SMMHC mainly activated their target genes in AMPs
To assess the role of Runx1 for Cbfb-MYH11–induced gene expression changes, we performed ChIC-seq assays31 on the AMP populations isolated from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice with RUNX1, CBFβ, SMMHC, H3K27ac, and H3K27me3 antibodies and identified their binding regions using the cLoops clustering algorithm.40 A significantly lower number of binding sites or genes assigned to the binding sites of RUNX1 and SMMHC was observed in the Runx1f/fMx1-CreCbfb+/56M samples than in the Mx1-CreCbfb+/56M samples (supplemental Figure 9A; supplemental Table 5). In contrast, the numbers of binding sites for CBFβ, H3K27ac, or H3K27me3 were not significantly changed between the 2 genotypes (supplemental Figure 9A). These findings suggested that RUNX1 was required for CBFβ-SMMHC, but not CBFβ, to interact with DNA targets.
Although the number of RUNX1 peaks in Runx1f/fMx1-CreCbfb+/56M samples was reduced by only 67% compared with that identified in Mx1-CreCbfb+/56M samples, most of the detected RUNX1 peaks (4279 of the total 5844 peaks) were observed in both sets of samples and the intensity of these peaks was much lower in the Runx1f/fMx1-CreCbfb+/56M samples than in the Mx1-CreCbfb+/56M samples (supplemental Figure 9B). The RUNX1 peaks unique to Runx1f/fMx1-CreCbfb+/56M samples were most likely identified because of the setting and high sensitivity of the peak-calling algorithm. As seen in supplemental Figure 9B, lower right panel, these peaks have much lower intensities than the other 2 types of peaks in supplemental Figure 9B, and they were not really unique to Runx1f/fMx1-CreCbfb+/56M samples because similar peaks were also identified in the Mx1-CreCbfb+/56M samples. These observations suggest that these RUNX1 peaks in the Runx1f/fMx1-CreCbfb+/56M samples were background/nonspecific binding of the RUNX1 antibody.
Among the RUNX1 binding sites identified in the AMP cells from mice of these 2 genotypes, ∼69.4% of them were only observed in Mx1-CreCbfb+/56M cells (supplemental Figure 9B). Of these binding sites unique to Mx1-CreCbfb+/56M cells, besides the RUNX (Runt) motif, GATA2 (GATA) motifs were also significantly enriched (supplemental Figure 9C), suggesting RUNX1 and GATA2 co-occupy many genomic regions to regulate the expression of nearby genes.25 All together, these results suggest that RUNX1 functions in part through GATA2 to regulate Cbfb-MYH11–induced leukemia.
As most DEGs (56.1%) between Runx1f/fMx1-CreCbfb+/56M and Mx1-CreCbfb+/56M mice, especially downregulated DEGs (72.7% vs 48.8% of upregulated DEG), were RUNX1 target genes (Figure 5A-B), we wondered how RUNX1 regulated the expression of these DEGs. We intersected the binding sites for RUNX1, CBFβ, and SMMHC with Intervene,41 and we considered the CBFβ and SMMHC co-occupied binding sites as CBFβ-SMMHC binding sites (the overlapped region between blue and green in supplemental Figure 9D). Interestingly, almost all such CBFβ-SMMHC binding sites (95.7%) were colocalized with RUNX1 binding sites, reinforcing the importance of RUNX1 for CBFβ-SMMHC in leukemogenesis. The RUNX1 and CBFβ-SMMHC co-occupied binding sites were then considered as RUNX1/CBFβ-SMMHC binding sites and genes assigned to these binding sites as target genes of RUNX1/CBFβ-SMMHC. Both upregulated and downregulated DEGs in Runx1f/fMx1-CreCbfb+/56M mice contained RUNX1/CBFβ-SMMHC target genes (Figure 5A,C). Interestingly, a significantly higher percentage of DEGs downregulated in Runx1f/fMx1-CreCbfb+/56M mice were RUNX1/CBFβ-SMMHC target genes than upregulated DEGs (21.5% vs 14.7%; Figure 5A,C), suggesting RUNX1/CBFβ-SMMHC was more responsible for the downregulated DEGs in Runx1f/fMx1-CreCbfb+/56M mice. Correspondingly, RUNX1 binding enrichment at the transcription start sites (TSSs) was reduced for both downregulated and upregulated DEGs in Runx1f/fMx1-CreCbfb+/56M cells (Figure 5D-E). On the other hand, reduced CBFβ and SMMHC bindings were only observed at TSSs of downregulated but not upregulated DEGs in Runx1f/fMx1-CreCbfb+/56M cells (Figure 5D-E). In addition, reduced H3K27ac modification was only observed at TSSs of downregulated DEGs in the absence of Runx1, which likely correlated with decreased expression of these genes in Runx1f/fMx1-CreCbfb+/56M cells. Conversely, reduced H3K27me3 modification was observed at TSSs of upregulated DEGs in the absence of Runx1, which likely correlated with increased expression of these genes in Runx1f/fMx1-CreCbfb+/56M cells (Figure 5D-E). Taken together, our results suggest that instead of acting as a transcription repressor, the transcription activation role of RUNX1/CBFβ-SMMHC is more prevalent in Cbfb-MYH11–induced leukemia.
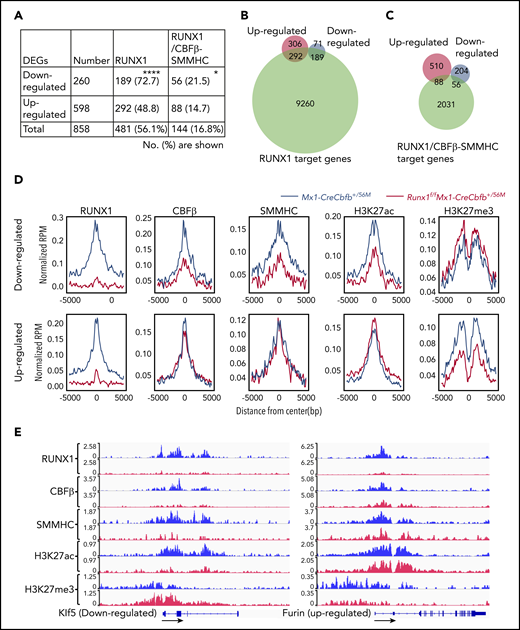
Target gene upregulation is a major function of CBFβ-SMMHC in leukemogenesis. (A-D) ChIC-seq was performed on AMP cells isolated from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice 2 to 3 weeks after pIpC treatment. (A) Number (percentage) of the downregulated or upregulated DEGs occupied by RUNX1 and RUNX1/CBFβ-SMMHC complex. *P < .05, ****P < .0001, between down- and upregulated DEGs by χ2 test. (B-C) Venn diagrams representing the overlap of DEGs between Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M AMPs (up- or downregulated in the Runx1f/fMx1-CreCbfb+/56M AMPs, same as below) and RUNX1 target genes (B) or RUNX1/CBFβ-SMMHC target genes (C) in AMPs from Mx1-CreCbfb+/56M cells. (D) Average binding profile of indicated proteins at the TSSs of the DEGs. (E) ChIC-seq analyses of RUNX1, CBFβ, SMMHC, H3K27ac and H3K27me3 binding at the Klf5 and Furin genes in AMP cells from Mx1-CreCbfb+/56M (blue) and Runx1f/fMx1-CreCbfb+/56M mice (red). Direction of the gene was indicated by black arrows.
Target gene upregulation is a major function of CBFβ-SMMHC in leukemogenesis. (A-D) ChIC-seq was performed on AMP cells isolated from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice 2 to 3 weeks after pIpC treatment. (A) Number (percentage) of the downregulated or upregulated DEGs occupied by RUNX1 and RUNX1/CBFβ-SMMHC complex. *P < .05, ****P < .0001, between down- and upregulated DEGs by χ2 test. (B-C) Venn diagrams representing the overlap of DEGs between Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M AMPs (up- or downregulated in the Runx1f/fMx1-CreCbfb+/56M AMPs, same as below) and RUNX1 target genes (B) or RUNX1/CBFβ-SMMHC target genes (C) in AMPs from Mx1-CreCbfb+/56M cells. (D) Average binding profile of indicated proteins at the TSSs of the DEGs. (E) ChIC-seq analyses of RUNX1, CBFβ, SMMHC, H3K27ac and H3K27me3 binding at the Klf5 and Furin genes in AMP cells from Mx1-CreCbfb+/56M (blue) and Runx1f/fMx1-CreCbfb+/56M mice (red). Direction of the gene was indicated by black arrows.
Single-cell RNA-seq identified the AMP population in the Runx1f/fMx1-CreCbfb+/56M mice as homogenous and distinct from that in the Mx1-CreCbfb+/56M mice
To explore the heterogeneity of the AMP population and determine which subpopulation was affected by Runx1 knockout, we performed single-cell RNA-sequencing (scRNA-seq) with AMP cells from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice. In the first set of experiment, 14 092 cells were captured, including 6456 cells from a Mx1-CreCbfb+/56M mouse and 7636 cells from a Runx1f/fMx1-CreCbfb+/56M mouse (supplemental Figure 10A). Clustering of pooled cells from both mice with uniform manifold approximation and projection42 allowed the separation of the cells into 4 clusters (Figure 6A), with most of the cells belonged to clusters 2 (42.05%) and 3 (54.3%). Further analysis revealed that the two smaller clusters, cluster 0 (1.11%) and cluster 1 (2.53%), contained neutrophil cells (expressing Elane and Mpo) and B cells (expressing Cd79A and Cd19), respectively (supplemental Figure 10B), which were not considered in subsequent data analyses. We then determined the distribution of the cells from those 2 mice. Interestingly, cluster 2 was mainly composed of cells from the Mx1-CreCbfb+/56M mouse and cluster 3 was mainly composed of cells from the Runx1f/fMx1-CreCbfb+/56M mouse (Figure 6B-C). These 2 clusters could not be subdivided further meaningfully, suggesting that the AMP cells were very homogenous in both mice. In the repeat experiment with another 2 mice, only 3 clusters were observed with 2 of them being the small contaminating B cells and neutrophils (Figure 6D; supplemental Figure 10B). Again, the main cluster (cluster 2 in Figure 6D) could be subdivided into 2 distinct, homogenous subclusters according to their genotypes, as observed in the first experiment (Figure 6E). Several genes were specifically expressed in Mx1-CreCbfb+/56M or Runx1f/fMx1-CreCbfb+/56M cells in both experiments (Figure 6C,F; supplemental Figure 10C; supplemental Table 6). The differences observed between these 2 experiments were most likely because of different mice used that were harvested at different times after pIpC injections. Taken together, these results suggest that the AMPs in the Runx1f/fMx1-CreCbfb+/56M mice are homogenous and distinct from those in the Mx1-CreCbfb+/56M mice at the transcriptome level, further confirms that Runx1 was required for the generation and maintenance of a functional AMP population.
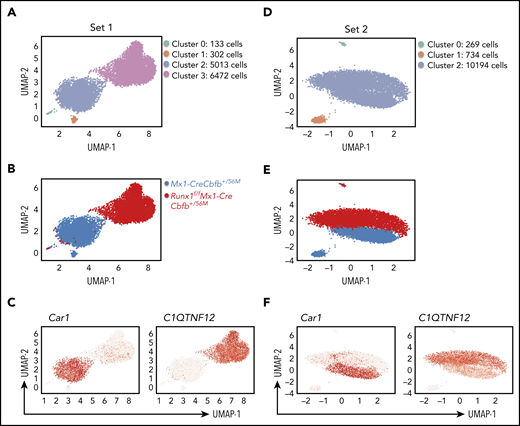
Runx1 is required for Cbfb-MYH11–induced generation of functional AMP population. Uniform manifold approximation and projection plots of AMP single cells from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice at 2 to 3 weeks after pIpC treatment. (A-C) the first set of experiments. (D-F) The second set of repeat experiments. (A,D) Clusters based on gene expression patterns. (B,E) Distributions of the cells of the 2 genotypes among the clusters. (C,F) Feature plot depicting the expression of genes (Car1 and C1QTNF12) across different cell populations (red, high; gray, low).
Runx1 is required for Cbfb-MYH11–induced generation of functional AMP population. Uniform manifold approximation and projection plots of AMP single cells from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice at 2 to 3 weeks after pIpC treatment. (A-C) the first set of experiments. (D-F) The second set of repeat experiments. (A,D) Clusters based on gene expression patterns. (B,E) Distributions of the cells of the 2 genotypes among the clusters. (C,F) Feature plot depicting the expression of genes (Car1 and C1QTNF12) across different cell populations (red, high; gray, low).
Discussion
Recent studies challenged the RUNX1 repression model and suggested a RUNX1 repression independent model for CBFβ-SMMHC–mediated leukemogenesis.18-22,24,25,30 Especially interesting was our previous finding that a moderate reduction of Runx1 activity (Runx1+/lz) delayed Cbfb-MYH11–induced leukemia,26 which suggested that not only CBFβ-SMMHC did not repress RUNX1, but it actually required RUNX1 for leukemogenesis. However, the mouse model used in our previous study was imperfect because the Runx1+/lz mice retained 1 wild-type Runx1 allele with normal RUNX1 function, in addition to an artificial Runx1-lz fusion allele.26 In this study, we performed the experiments with an inducible homozygous Runx1 knockout model, which should retain no functional RUNX1 (Figure 1).
Strikingly, leukemia did not develop in any mice with Runx1 knockout and Cbfb-MYH11 knockin (Runx1f/fMx1-CreCbfb+/56M; Figure 2A; supplemental Figure 5), indicating that RUNX1 was required for leukemogenesis by CBFβ-SMMHC. We also found that the AMPs, a population unique to Cbfb-MYH11–expressing mice and able to induce leukemia,28 decreased and then disappeared in Runx1f/fMx1-CreCbfb+/56M mice shortly after pIpC treatment (Figure 3A,C,D; supplemental Figure 7B). These results suggest that leukemogenesis from these AMPs requires RUNX1. It is possible that other cell populations in the bone marrow, especially hematopoietic stem cells and multipotent progenitors were also affected by RUNX1 loss; however, we decided to focused on the AMPs because of their dynamic change in the Runx1f/fMx1-CreCbfb+/56M mice and the fact that they are considered as the leukemia-initiating cells.
To address how Runx1 cooperated with Cbfb-MYH11 for leukemogenesis, we first performed bulk RNA-seq of the AMPs from Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M mice to determine the effect of Runx1 knockout on transcriptome. We found that genes associated with proliferation, differentiation blockage, and maintenance of leukemia-initiating ability were differentially expressed between Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M AMPs (Figure 4), suggesting these genes were required for the generation and function of the AMPs. We also performed ChIC-seq to determine the effect of Runx1 knockout on target gene binding and histone modifications. Results from the ChIC-seq assay imply that RUNX1 is required for the recruitment of CBFβ-SMMHC to target genes in AMPs and that RUNX1 and CBFβ-SMMHC work together to both activate and inhibit, especially activate, target genes expression. Of the DEGs between Mx1-CreCbfb+/56M and Runx1f/fMx1-CreCbfb+/56M AMPs, a significantly higher percentage of DEGs downregulated in Runx1f/fMx1-CreCbfb+/56M AMPs were RUNX1/CBFβ-SMMHC target genes compared with upregulated DEGs. In addition, CBFβ and SMMHC bindings were reduced at TSSs only for DEGs downregulated in Runx1f/fMx1-CreCbfb+/56M cells, although RUNX1 binding was reduced at TSSs of both downregulated and upregulated DEGs in Runx1f/fMx1-CreCbfb+/56M cells. As expected, reduced H3K27ac modification was observed at TSS of downregulated DEGs in the Runx1f/fMx1-CreCbfb+/56M cells, correlating with decreased expression of these genes. Together, the RNA-seq and ChIC-seq data demonstrate that RUNX1 and CBFβ-SMMHC more frequently function as a transcription activator complex and possibly recruit other proteins, such a GATA2, histone acetyl transferases and/or histone deacetylases10 to promoters of many target genes, inducing H3K27ac modification and eventually leading to enhanced genes expression. These results are consistent with previous findings that most of the gene expression changes induced by CBFβ-SMMHC are increases in expression.25,43
Interestingly, 30% of Runx1f/fMx1-CreCbfb+/56M mice, in both primary and transplant models, died around 2 weeks after pIpC treatment with no signs of leukemia, but multilineage pancytopenia with especially severe thrombocytopenia and anemia, which were not observed in Mx1-CreCbfb+/56M or Runx1f/fMx1-Cre mice. The exact cause of death was not clear, probably because of a combined effect of genetic changes (loss of Runx1 and expression of Cbfb-MYH11) and pIpC treatment. Runx1 is known to be important for megakaryocytic differentiation while pIpC treatment can suppress erythroid development,44 and it may induce cell death through its effector, type I interferons.45 Although beyond the scope of this paper, it will be interesting to try another Cre system, such as tamoxifen-inducible CreER, to determine how important pIpC treatment is for this early lethality phenotype in the Runx1f/fMx1-CreCbfb+/56M mice.
In this study, we show that Runx1 is required for Cbfb-MYH11–induced leukemogenesis. In addition, we provide important insight into the mechanism of leukemogenesis associated with CBFβ-SMMHC (ie, the fusion protein acts as a transactivator of gene expression in the presence of RUNX1). These results therefore validate current efforts to develop inhibitors of RUNX1–CBFβ-SMMHC interaction for the treatment of inv(16) AML.46,47
For original data, please contact pliu@nih.gov. The data from RNA-seq, ChIC-seq and single cell RNA-sequencing have been deposited in GEO with accession no. GSE152573.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Irene C. Ginty, Weiwei Wu, Abdel G. Elkahloun, Stephen Wincovitch, Stacy Anderson, Martha Kirby, and the National Institutes of Health (NIH) Intramural Sequencing Center for technical help. This work used the computational resources of the NIH high-performance computing Biowulf cluster (https://hpc.nih.gov).
The research was supported by the Intramural Research Programs of National Human Genome Research Institute and National Heart, Lung, and Blood Institute, National Institutes of Health.
Authorship
Contribution: T.Z. designed and performed experiments, analyzed data, and wrote the paper; Y.C. analyzed the bulk RNA-seq, ChIC-seq, and scRNA-seq data and wrote the paper; G.R., L.Z., R.K.H., G.L., D.F., and L.A. performed experiments; K.Z. contributed to the ChIC-seq experimental design and data analysis; and P.P.L. designed the experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no financial competing interests.
Correspondence: P. Paul Liu, 50 South Dr, Bldg 50, Rm 5222C, NHGRI, NIH, Bethesda, MD 20892; e-mail: pliu@mail.nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal