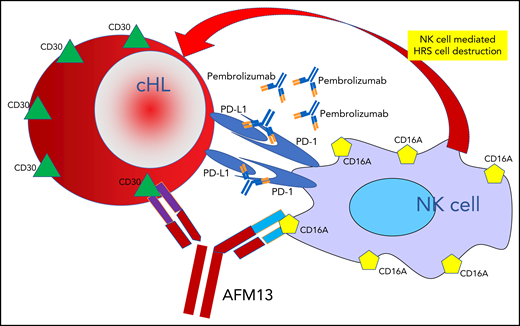
In their article in this issue of Blood describing the use of a new bispecific antibody, AFM13, plus a checkpoint inhibitor, pembrolizumab, for patients with recurrent or refractory classic Hodgkin lymphoma (cHL), provide dramatic evidence of the treatment potency that can be achieved with a 3-pronged therapeutic attack employing 2 biologic agents targeting specific immunologic characteristics of cHL Hodgkin and Reed-Sternberg (HRS) cells.1 They show how to forcefully recruit the patient’s immune system’s killer cells while at the same time enhancing their cytodestructive potency. Their work nicely demonstrates how recent progress in the treatment of cHL has been carefully built on an increasingly exquisite understanding of the molecular biology underlying the immunology of malignant HRS cells and their dynamic interaction with both specific and nonspecific effector cells in the immune system.
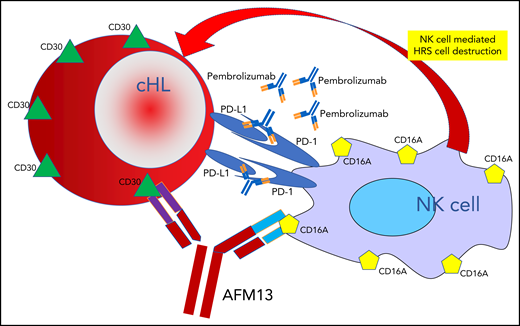
One arm of the bispecific antibody AFM13 binds to CD30 surface antigens on cHL HRS cells, and the other arm binds to CD16A surface antigens on NK cells, leading to NK cell–mediated destruction of the HRS cells. This therapeutic attack is dramatically enhanced by adding pembrolizumab, which blocks the protection from immune attack that HRS cells employ by elaborating PD-L1.
One arm of the bispecific antibody AFM13 binds to CD30 surface antigens on cHL HRS cells, and the other arm binds to CD16A surface antigens on NK cells, leading to NK cell–mediated destruction of the HRS cells. This therapeutic attack is dramatically enhanced by adding pembrolizumab, which blocks the protection from immune attack that HRS cells employ by elaborating PD-L1.
The universal, persistent expression of the CD30 antigen on the surface of cHL cells provides an attractive target for specially crafted antibodies, a property that has been effectively exploited by the development of the antibody-drug conjugate brentuximab vedotin, which employs a CD30 targeted antibody to deliver the microtubule disrupting effector toxin monomethyl aurostatin E to the target HRS cells.2,3 The novel bispecific antibody AFM13 targets that same CD30 antibody with 1 arm of a monoclonal antibody, while at the same time, the other arm binds to the CD16A cell surface antigen expressed on natural killer (NK) cells (see figure), thus, dragging the NK cells into tight proximity with HRS cells. Once there, however, the NK cells have only limited capacity to injure the HRS cells, presumably at least in part because the HRS cells express program death-ligand 1 (PD-L1), which blunts the NK cell’s ability to disrupt the HRS cells, accounting for the limited activity of AFM13 as a single agent (overall and partial response rates only 12%).4 By adding pembrolizumab, a checkpoint inhibitor, to the treatment mix, Bartlett and her coinvestigators cleverly overcome this NK cell blunting, releasing the destructive potential of the NK cells, resulting in impressive overall and complete response rates of 88% and 42%, respectively, in 30 patients with advanced, recurrent, or refractory cHL that had proven resistant to standard therapy, including brentuximab vedotin in 43% and autologous hematopoietic stem cell transplant in 40% of the patients. Gratifyingly, reflecting the biologic specificity of the treatments and resultant ability to avoid off target toxicity, this therapeutic potency is achieved with modest toxicity, primarily consisting of infusion reactions, reaching grade 3 in only 13% of patients.
Progress in the treatment of advanced stage cHL has unfolded in 3 major therapeutic eras. First, multidrug combinations of cytotoxic chemotherapeutic agents, such as ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine), demonstrated that even far advanced disease can be cured.5,6 Next, highly intensified chemotherapy was shown to be able to increase cure rates either when employed for disease in relapse despite primary chemotherapy7,8 or in first-line treatments, such as escalated BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, prednisone, and procarbazine).9 In both eras increased efficacy came with the high price of increased short- and long-term toxicity. Now, in the third major era, that of active immunologic treatment of cHL, additional progress is being achieved by exploiting insights into the basic biology of HRS cells and their complex interaction with immunologic effector cells in the tumor microenvironment. Results achieved with checkpoint inhibitors, such as nivolumab and pembrolizumab, which target and block the inhibition of immune cell destruction of HRS cells by cytotoxic effector cells induced when HRS cells produce programmed death ligands, document the high level of effectiveness that can be achieved by unleashing immune effector cells to attack HRS cells. A remaining obstacle to further progress, however, consisted of how to bring the effector NK cells into close proximity with the target HRS cells and then, once these cells are in position, how to overcome the inhibition posed by HRS cells’ innate ability to block destruction of target cells by elaborating programmed death inhibitory molecules. The AFM13 investigators settled on the clever 3-pronged approach of using a bispecific antibody, AFM13, to pull NK cells into tight proximity with HRS cells and at the same time flooding the system with pembrolizumab, to eliminate the programmed cell death-1/PD-L1 enforced blockage of NK cell–mediated target cell destruction (see figure). This approach works nicely, producing clinically useful responses in most patients, including complete metabolic responses in almost half, and does so with minimal toxicity, a direct consequence of employing biologically selective agents.
The full potential that can be realized in this third major therapeutic era of the treatment of cHL will only be reached when novel immunologic approaches such as AFM13 plus a checkpoint inhibitor are integrated into front-line treatment. Guided and encouraged by the evidence for a high level of efficacy provided by the AFM13 investigators, clinical trials can now be launched employing these biologically selective therapies in earlier and eventually front-line clinical trials. Much remains to be achieved, but the treatment of cHL with immunologically selective, largely nontoxic treatments is now well underway.
Conflict-of-interest disclosure: The author declares no competing financial interests.