

Key Points
AFM13, a bispecific, tetravalent innate cell engager, activates natural killer cells and macrophages via CD16A to target CD30+ lymphoma cells.
AFM13 in combination with pembrolizumab for HL patients was well-tolerated with adverse events that were generally manageable.

Abstract
In relapsed/refractory Hodgkin lymphoma (R/R HL), immunotherapies such as the anti-programmed death-1 inhibitor pembrolizumab have demonstrated efficacy as monotherapy and are playing an increasingly prominent role in treatment. The CD30/CD16A-bispecific antibody AFM13 is an innate immune cell engager, a first-in-class, tetravalent antibody, designed to create a bridge between CD30 on HL cells and the CD16A receptor on natural killer cells and macrophages, to induce tumor cell killing. Early studies of AFM13 have demonstrated signs of efficacy as monotherapy for patients with R/R HL and the combination of AFM13 with pembrolizumab represents a rational new treatment modality. Here, we describe a phase 1b, dose-escalation study to assess the safety and preliminary efficacy of AFM13 in combination with pembrolizumab in patients with R/R HL. The primary objective was estimating the maximum tolerated dose; the secondary objectives were to assess safety, tolerability, antitumor efficacy, pharmacokinetics, and pharmacodynamics. In this heavily pretreated patient population, treatment with the combination of AFM13 and pembrolizumab was generally well tolerated, with similar safety profiles compared to the known profiles of each agent alone. The combination of AFM13 with pembrolizumab demonstrated an objective response rate of 88% at the highest treatment dose, with an 83% overall response rate for the overall population. Pharmacokinetic assessment of AFM13 in the combination setting revealed a half-life of up to 20.6 hours. This proof-of-concept study holds promise as a novel immunotherapy combination worthy of further investigation. This phase 1b study was registered at www.clinicaltrials.gov as NCT02665650.
Introduction
Patients with Hodgkin lymphoma (HL) have the potential to be cured or experience long-term remission with risk-adapted treatment, including chemotherapy and radiotherapy, but 10% to 30% can develop progressive disease or relapse.1 For patients with relapsed or refractory HL, 50% or fewer can be cured with high-dose chemotherapy and autologous stem cell transplantation (ASCT).2,3 Historically, patients with HL who relapse or progress after ASCT have a poor prognosis with a median overall survival of ∼2 years; however, outcomes have improved substantially with the development of new drug classes.4,5 A recent retrospective study reported that, for patients treated with novel agents (excluding immune checkpoint inhibitors) after post-ASCT relapse, the median overall survival was 85.6 months.6,7
Brentuximab vedotin (BV) was the first targeted therapy to be approved (in the United States) for the treatment of HL, indicated for use in the R/R setting. BV is an antibody drug conjugate (ADC) that targets CD30 and is conjugated to a cytotoxic agent. A pivotal phase 2 study reported that BV treatment resulted in an overall response rate (ORR) of 75% in patients with relapsed/refractory (R/R) HL and a complete response (CR) rate of 35%. However, the median progression-free survival after BV treatment is only 5.7 months, and treatment-emergent adverse events such as grade 3/4 neutropenia and neuropathy are common.8
Immunotherapy is a promising new treatment option for HL. Anti-programmed death-1 (PD-1) antibodies pembrolizumab and nivolumab have produced striking results in patients with relapsed or refractory disease as monotherapy with an ORR of 69% for both pembrolizumab and nivolumab.9,10 Both antibodies have been well tolerated with CRs in the range of 22.4% and 16%, respectively. For HL, therapeutic PD-1 blockade is largely independent of major histocompatibility complex class I (MHC-I)-mediated CD8+ T-cell responses, whereas MHC II expression on HL cells was predictive for complete remission, suggesting CD4+ T cells can play a therapeutic role.11 Targeted ADCs and immunotherapies represent promising approaches for the treatment of R/R HL. However, additional therapeutic options and combination therapy are needed with greater and more durable CR rates with favorable or manageable toxicity profiles.
AFM13, a first-in-class innate cell engager, is in clinical development for treatment of CD30+ lymphomas including R/R HL and peripheral T-cell lymphoma. Developed by the fit-for-purpose ROCK platform that generates customizable antibodies, AFM13 is a CD16A/CD30 tetravalent, bispecific antibody stimulating innate immune cells, such as natural killer (NK) cells and macrophages.12,13 AFM13 binds CD16A on innate cells and binds CD30 on HL cells, acting as a bridge to recruit and activate innate immune cells in close proximity to tumor cells.14-16 The activating receptor CD16A on NK cells facilitates antibody-dependent cell-mediated cytotoxicity (ADCC) and is the only activating receptor triggering the cytotoxic activity of naïve human NK cells.15 Research suggests macrophages are also engaged by AFM13, contributing to the innate immune response.17 AFM13, as the most clinically advanced innate immune cell engager, was first studied in HL patients as monotherapy in a dose-escalating phase 1 clinical study for patients with R/R HL.18 AFM13 treatment was safe, well tolerated, and resulted in objective tumor responses in multiple patients.18 In this study, AFM13 demonstrated significant NK cell activation and a decrease of soluble CD30 in peripheral blood as well as activity in HL patients who received prior BV.18 Phenotypic analysis of lymphoma cells from patients with HL, refractory to or relapsed after treatment with BV, show that CD30 expression levels are sustained, providing rationale for targeting CD30+ lymphoma with AFM13 in this setting.19 We present the results of an open-label, multicenter, phase 1b, dose-escalation study to assess the safety and efficacy of AFM13 in combination with pembrolizumab in patients with R/R HL.
Patients, materials, and methods
Patients
This study included patients with CD30+ classical HL confirmed by histopathology who were R/R after standard therapy, including BV. Where applicable, patients were required to have completed ASCT ≥3 months before the first study dose. Further inclusion criteria were ≥18 years of age, Eastern Cooperative Oncology Group performance status <2, and signed informed consent. Patients were excluded if they had received prior therapy with an anti-PD-1, anti-PD-L1, or anti-PD-L2 agent. Patients having certain diseases (other than HL), such as history of interstitial lung disease, evidence of current central nervous system involvement, immunodeficiency disorders, or ongoing systemic corticosteroid treatment, were excluded.
Study design and procedures
This open-label, multicenter, phase 1b, dose-escalation study with extension cohort was conducted at 14 US sites and 4 sites in Spain from June 2016 to March 2019. The primary objective of this study was to estimate the maximum tolerated dose (MTD)/maximum administered dose (MAD) of the combination of AFM13 and pembrolizumab. The study consisted of 2 parts; part 1 (primary cohorts) followed a 3+3 study design in which doses of AFM13 were escalated in sequential cohorts, whereas the dose of pembrolizumab was fixed at 200 mg given every 3 weeks in all patients. If 2 or more dose-limiting toxicity (DLT) events occurred in an expanded cohort, the next lower dose level was assigned as the MTD. If the maximum dose of AFM13 (3.0/7.0 mg/kg) was reached without need to deescalate because of DLT, it was defined as the MAD. An extension cohort (part 2) allowed additional evaluation of the MTD or MAD of AFM13 when given in combination with pembrolizumab. Patients were recruited to the extension cohort with the intention to assess up to 21 evaluable patients with the selected combination.
Secondary objectives were to assess the safety and tolerability of increasing doses of AFM13 in combination with pembrolizumab, to assess antitumor activity of AFM13 in combination with pembrolizumab, and to evaluate the pharmacokinetic profile of AFM13 when combined with pembrolizumab. Exploratory analyses included the assessment of relevant biomarkers and to assess the immunogenicity of AFM13.
The dosing of pembrolizumab was started in week 1 and AFM13 in week 2 (Table 1; supplemental Figure 1). Patients were assigned to 1 of 3 cohorts (part 1) and received AFM13 IV over 2 or 4 hours (for doses ≤3 mg/kg and >3 mg/kg, respectively): cohort 1: 0.1 mg/kg 3 times per week for up to 2 weeks (ie, week 2 to week 3), 0.5 mg/kg per week for up to 6 weeks (week 4 to week 9), and 0.5 mg/kg every 3 weeks for up to 16 weeks (week 10 to week 25); cohort 2: 0.5 mg/kg 3 times per week for up to 2 weeks, 1.5 mg/kg/week for up to 6 weeks, and 1.5 mg/kg every 3 weeks for up to 16 weeks; cohort 3: 3.0 mg/kg 3 times per week for up to 2 weeks, 7.0 mg/kg per week for up to 6 weeks, and 7.0 mg/kg every 3 weeks for up to 16 weeks. The 7.0 mg/kg dose level was used in the clinical development of AFM13 when given as monotherapy; it was not planned to escalate beyond this. All patients received 200 mg of pembrolizumab every 3 weeks until disease progression, unacceptable toxicity, cessation of treatment because of CR (at investigator’s discretion), or 52 weeks of treatment, whichever occurred first.
Dosing schedule for AFM13 and pembrolizumab as combination therapy
| AFM13 dose levels (mg/kg) | Pembrolizumab (mg) | |||
|---|---|---|---|---|
| Weeks 2 and 3 | Weeks 4-9 | Weeks 10, 13, 16, 19, 22, 25 | Weeks 1-52 Q3W* | |
| Cohort 1 | 0.1 × 3 | 0.5 | 0.5 | 200 |
| Cohort 2 | 0.5 × 3 | 1.5 | 1.5 | 200 |
| Cohort 3 | 3.0 × 3 | 7.0 | 7.0 | 200 |
| AFM13 dose levels (mg/kg) | Pembrolizumab (mg) | |||
|---|---|---|---|---|
| Weeks 2 and 3 | Weeks 4-9 | Weeks 10, 13, 16, 19, 22, 25 | Weeks 1-52 Q3W* | |
| Cohort 1 | 0.1 × 3 | 0.5 | 0.5 | 200 |
| Cohort 2 | 0.5 × 3 | 1.5 | 1.5 | 200 |
| Cohort 3 | 3.0 × 3 | 7.0 | 7.0 | 200 |
Q3W, once every 3 weeks.
Until disease progression, unacceptable toxicity, cessation of treatment because of complete response (at investigator’s discretion) or 52 weeks of treatment, whichever occurred first.
Study oversight
The protocol was approved by the institutional review board at each center, and the study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. All the patients provided written informed consent before study entry. The principal investigators, in collaboration with the sponsor (Affimed GmbH), were responsible for the design and oversight of the study and development of the protocol, available at ashpublications.org/blood. The sponsor was responsible for the collection and maintenance of the data. The manuscript was drafted by a third party (W2O), paid for by the sponsor, and all authors reviewed and approved the draft manuscript. All the authors made the decision to submit the manuscript for publication and attest for the accuracy and completeness of the data reported and adherence to the protocol.
Study assessments
Safety was assessed by Common Terminology Criteria for Adverse Events, version 4.03, and included clinical examinations, the assessment of adverse events (AE), DLT, and laboratory parameters. AE were assessed at each visit, in addition to concomitant medications and laboratory screenings. A treatment-emergent adverse event was defined as an adverse event with an onset date on or after the first dose of study drugs, and on or before 30 days after the last dose of study drugs. Patients on the study were treated with H1/H2 antagonists as a required premedication. After the occurrence of 2 ≥grade 3 infusion-related reaction (IRR) events, the protocol was amended to add acetaminophen and corticosteroid to the premedication regimen.
AFM13 was only administered from week 2 through week 25, whereas pembrolizumab continued as a single agent through week 52 (supplemental Figure 1). In addition, study drug infusions were administered with a safety observation period in between, and the investigators were allowed to attribute events to a single drug or the combination therapy. During the safety assessment and review, each event was accounted for only once to avoid duplication.
Tumor response was assessed by investigators according to the revised response criteria for malignant lymphoma (Lugano classification according to the Cheson criteria, 2014) and by an independent data monitoring reviewer.20 Tumor imaging (18F fluorodeoxyglucose positron emission tomography and computed tomography) was performed on all patients at screening and after every 12 weeks. Tumor response was determined by the investigator on computed tomography-based radiographic response and 18F fluorodeoxyglucose positron emission tomography-based metabolic response separately. Blood samples to measure levels of soluble CD30 in serum were taken at screening; weeks 7, 13, and 25; and the final visit. Samples for assessment of anti-drug antibodies (ADA) against AFM13 were taken at screening, week 7, and at week 25 or the final visit, whichever occurred first. Where possible, patients were followed up every 3 months from their final visit to check for disease progression and/or survival status.
All participants in the extension cohort (part 2) participated in an analysis where their predose AFM13 serum trough levels were assessed for each AFM13 infusion. Four patients in part 2 had blood samples taken to assess the pharmacokinetic profile of AFM13 around their first infusion in week 2 (day 8), after the last infusion in week 3 (day 19/20), and after the infusion in week 7. Up to 12 patients enrolled in part 2 who provided additional informed consent for the translational substudy had 2 biopsies and additional blood samples taken (group 1 at screening and before the first pembrolizumab/AFM13 same-day dosing at week 4, and group 2 at screening and before the second pembrolizumab dose at week 1).
Pharmacokinetic parameters assessed included maximum serum concentration, area under the curve, volume of distribution, half-life, and systemic clearance.
Data evaluation
Two populations for analysis were considered. The dose-determining set consisted of all patients in each cohort who completed 6 weeks of treatment and patients who discontinued earlier because of a DLT occurring in this period. This population was used for the dose escalation in part 1. For all other objectives, the safety set consisted of all patients that received at least 1 dose of AMF13 or pembrolizumab. Patients were classified in actual-treatment-received cohorts. There was no inferential statistical analysis for primary end points in this study. Results were listed and summarized using descriptive statistics. For all secondary and exploratory objectives, results were listed and summarized using descriptive statistics.
Results
Patient characteristics
A total of 30 patients were enrolled in this phase 1b, dose-escalation study (N = 30). The median age (range) was 34 years (18-73 years) and the majority (67%) were male. This was a heavily pretreated patient population (3-7 prior therapies) with 14/30 patients (46.7%) having 3 prior therapies. A total of 12 patients (40%) had a prior ASCT. All 30 patients had relapsed or refractory disease (43% relapsed, 57% refractory), and 43% of patients (13/30) had BV as their last prior therapy (Table 2).
Subject demographics and baseline characteristics
| Part 1 | Part 2 | |||||
|---|---|---|---|---|---|---|
| Cohort 1 (0.1 × 3)/0.5 mg/kg (n = 3) | Cohort 2 (0.5 × 3)/1.5 mg/kg (n = 3) | Cohort 3 (3.0 × 3)/7.0 mg/kg (n = 6) | (3.0 × 3)/ 7.0 mg/kg (n = 18) | MAD* (3.0 × 3)/ 7.0 mg/kg (n = 24) | All patients (N = 30) | |
| Median age, y (min, max) | 29.0 (25, 73) | 34.0 (33, 53) | 36.0 (26, 49) | 27.5 (18, 52) | 32.0 (18, 52) | 33.5 (18, 73) |
| Sex | ||||||
| Male | 2 (66.7) | 2 (66.7) | 5 (83.3) | 11 (61.1) | 16 (66.7) | 20 (66.7) |
| Female | 1 (33.3) | 1 (33.3) | 1 (16.7) | 7 (38.9) | 8 (33.3) | 10 (33.3) |
| Prior therapies, no. (%) | ||||||
| 3 | 0 | 0 | 0 | 14 (77.8) | 14 (58.3) | 14 (46.7) |
| 4 | 1 (33.3) | 1 (33.3) | 3 (50.0) | 2 (11.1) | 5 (20.8) | 7 (23.3) |
| 5 | 0 | 1 (33.3) | 2 (33.3) | 0 | 2 (8.3) | 3 (10.0) |
| 6 | 1 (33.3) | 1 (33.3) | 1 (16.7) | 1 (5.6) | 2 (8.3) | 4 (13.3) |
| 7 | 1 (33.3) | 0 | 0 | 1 (5.6) | 1 (4.2) | 2 (6.7) |
| Prior autologous stem cell transplant | 2 (66.7) | 3 (100.0) | 4 (66.7) | 3 (16.7) | 7 (29.2) | 12 (40.0) |
| Part 1 | Part 2 | |||||
|---|---|---|---|---|---|---|
| Cohort 1 (0.1 × 3)/0.5 mg/kg (n = 3) | Cohort 2 (0.5 × 3)/1.5 mg/kg (n = 3) | Cohort 3 (3.0 × 3)/7.0 mg/kg (n = 6) | (3.0 × 3)/ 7.0 mg/kg (n = 18) | MAD* (3.0 × 3)/ 7.0 mg/kg (n = 24) | All patients (N = 30) | |
| Median age, y (min, max) | 29.0 (25, 73) | 34.0 (33, 53) | 36.0 (26, 49) | 27.5 (18, 52) | 32.0 (18, 52) | 33.5 (18, 73) |
| Sex | ||||||
| Male | 2 (66.7) | 2 (66.7) | 5 (83.3) | 11 (61.1) | 16 (66.7) | 20 (66.7) |
| Female | 1 (33.3) | 1 (33.3) | 1 (16.7) | 7 (38.9) | 8 (33.3) | 10 (33.3) |
| Prior therapies, no. (%) | ||||||
| 3 | 0 | 0 | 0 | 14 (77.8) | 14 (58.3) | 14 (46.7) |
| 4 | 1 (33.3) | 1 (33.3) | 3 (50.0) | 2 (11.1) | 5 (20.8) | 7 (23.3) |
| 5 | 0 | 1 (33.3) | 2 (33.3) | 0 | 2 (8.3) | 3 (10.0) |
| 6 | 1 (33.3) | 1 (33.3) | 1 (16.7) | 1 (5.6) | 2 (8.3) | 4 (13.3) |
| 7 | 1 (33.3) | 0 | 0 | 1 (5.6) | 1 (4.2) | 2 (6.7) |
| Prior autologous stem cell transplant | 2 (66.7) | 3 (100.0) | 4 (66.7) | 3 (16.7) | 7 (29.2) | 12 (40.0) |
MTD was not reached.
Safety
All 30 patients completed the 6-week DLT observation period. Twelve patients were treated in the dose escalation cohorts (cohorts 1, 2, and 3) and 18 in the extension cohort (same dose/schedule as cohort 3). One DLT was observed in a patient in cohort 3 (missing ≥25% of AFM13 dose during the DLT period because of grade 2 IRR), and another DLT was observed in a patient in the extension cohort (grade 4 IRR). The MTD of AFM13 was not exceeded in combination with pembrolizumab and the MAD of AFM13 was determined to be 7 mg/kg. The most common (≥10%) treatment-related AE for AFM13 were IRR (90%), rash (30%), nausea or pyrexia (23% each), diarrhea (20%), fatigue or headache (17% each), and increased aspartate aminotransferase (AST) or increased alanine aminotransferase (ALT) (13% each) (Table 3). The most common treatment-related AE of grade ≥3 for AFM13 included IRR (4 patients [13%]), and 1 patient (3%) each with gastritis, hypotension, increased AST, nausea, neutropenia, or vomiting. Most AE were low grade in nature and manageable with standard-of-care therapies. The most common treatment-related AE for AFM13 and pembrolizumab in combination (≥10%) included IRR (8 patients [27%]), rash (6 patients [20%]), nausea, (7 patients [23%]), fatigue and diarrhea (5 patients [17%] each), and headache and elevated ALT (3 patients [10%] each). Treatment-related AE of grade ≥3 for AFM13 and pembrolizumab in combination included 1 patient (3%) for each of the following: IRR, nausea, gastritis, and vomiting (Table 3).
Adverse events related to AFM13 and combination treatment, respectively
| AFM13 | AFM13 + pembrolizumab | |||
|---|---|---|---|---|
| All grades, ≥10% (n = 30), n (%) | ≥Grade 3 (n = 30), n (%) | All grades, ≥10%, n (%) | ≥Grade 3 (n = 30), n (%) | |
| Any AE | 29 (97) | 7 (23) | 22 (73) | 2 (7) |
| IRR | 27 (90) | 4 (13) | 8 (27) | 1 (3) |
| Rash | 9 (30) | — | 6 (20) | — |
| Nausea | 7 (23) | 1 (3) | 7 (23) | 1 (3) |
| Pyrexia | 7 (23) | — | 4 (13) | — |
| Fatigue | 5 (17) | — | 5 (17) | — |
| Diarrhea | 6 (20) | — | 5 (17) | — |
| Headache | 5 (17) | — | 3 (10) | — |
| Elevated ALT | 4 (13) | — | 3 (10) | — |
| Elevated AST | 4 (13) | 1 (3) | 2 (7) | — |
| Neutropenia | 2 (7) | 1 (3) | 2 (7) | — |
| Gastritis | 1 (3) | 1 (3) | 1 (3) | 1 (3) |
| Vomiting | 2 (7) | 1 (3) | 1 (3) | 1 (3) |
| Hypotension | 1 (3) | 1 (3) | — | — |
| Thrombocytopenia | 2 (7) | — | 2 (7) | — |
| URTI | 2 (7) | — | 2 (7) | — |
| AFM13 | AFM13 + pembrolizumab | |||
|---|---|---|---|---|
| All grades, ≥10% (n = 30), n (%) | ≥Grade 3 (n = 30), n (%) | All grades, ≥10%, n (%) | ≥Grade 3 (n = 30), n (%) | |
| Any AE | 29 (97) | 7 (23) | 22 (73) | 2 (7) |
| IRR | 27 (90) | 4 (13) | 8 (27) | 1 (3) |
| Rash | 9 (30) | — | 6 (20) | — |
| Nausea | 7 (23) | 1 (3) | 7 (23) | 1 (3) |
| Pyrexia | 7 (23) | — | 4 (13) | — |
| Fatigue | 5 (17) | — | 5 (17) | — |
| Diarrhea | 6 (20) | — | 5 (17) | — |
| Headache | 5 (17) | — | 3 (10) | — |
| Elevated ALT | 4 (13) | — | 3 (10) | — |
| Elevated AST | 4 (13) | 1 (3) | 2 (7) | — |
| Neutropenia | 2 (7) | 1 (3) | 2 (7) | — |
| Gastritis | 1 (3) | 1 (3) | 1 (3) | 1 (3) |
| Vomiting | 2 (7) | 1 (3) | 1 (3) | 1 (3) |
| Hypotension | 1 (3) | 1 (3) | — | — |
| Thrombocytopenia | 2 (7) | — | 2 (7) | — |
| URTI | 2 (7) | — | 2 (7) | — |
URTI, upper respiratory tract infection.
No AFM13 dose modifications were observed in cohorts 1 and 2, whereas a total of 7 patients required at least 1 AFM13 dose modification in cohort 3 (4 patients, total n = 6) and part 2 (3 patients, total n = 18). AFM13 dose interruptions were reported in 90% of patients from all cohorts (27 patients, N = 30). A total of 6 patients experienced AE leading to any study-drug discontinuation (5 AFM13 only, 1 AFM13 and pembrolizumab) and were reported as IRR (n = 5), acute myocardial infarction (n = 1), myocardial ischemia (n = 1), gastritis (n = 1), and hypotension (n = 1). Both events (acute myocardial infarction and myocardial ischemia) occurred in the same patient with multiple cardiovascular risk factors. All discontinuations were deemed related to study treatment(s). There was no DLT-related AFM13 discontinuation. A review of the dose exposure does not suggest any cumulative toxicity in patients as a result of prolonged dosing. The median dosing durations were 15.7 and 21.3 weeks for cohort 3 and part 2, respectively, and the median dose intensities were 5.2 and 4.3 mg/kg/week, respectively. These findings do not correlate with a higher frequency of AEs in cohort 3 compared with part 2 of the study.
There were 4 deaths during the course of the study, 1 case from a nontreatment-emergent event of vanishing bile duct syndrome and Steven Johnson syndrome (n = 1); the remaining cases were due to Aspergillus pneumonia and multiorgan failure (n = 1), mycosis fungoides–associated skin infection, septic shock, and cardiac arrest (n = 1), and respiratory insufficiency and pulmonary lymphoma (n = 1); the latter 3 deaths occurred in the context of progressive disease.
AFM13 trough levels rapidly reached steady state over the first 2 weeks (3 mg/kg 3 times per week); during the subsequent 7 mg/kg weekly dosing period, AFM13 trough concentrations remained constant, although at a lower level as observed over the first 2 weeks during the subsequent 7 mg/kg weekly dosing period. The apparent volume of distribution of up to 3.07 L was small, indicating that AFM13 is mainly confined to the plasma. The apparent terminal half-life ranged from 8.7 to 20.6 hours. Following IV dosing of AFM13 at doses of 3 to 7 mg/kg coadministered with 200 mg pembrolizumab in this combination trial, the apparent terminal half-life of AFM13 appears similar in the presence or absence of pembrolizumab.18 After AFM13 treatment initiation, ADA were detected in 17 of 30 patients. One patient had detectable ADA only at screening. Preliminary analysis of the data shows that there is no relationship between the development of ADA and AFM13 pharmacokinetics. Soluble CD30 levels were significantly reduced in the majority of the patients across all dose cohorts on week 7 and remained below baseline level at week 13.
Response to treatment
All 30 patients were evaluable for efficacy. The ORR was 83% (25 patients) by investigator assessment; 37% (11 patients) experienced a complete metabolic response (CMR) and 47% (14 patients) experienced a partial metabolic response (PMR) (Table 4). Seven percent (2 patients) showed no metabolic response (NMR), and 10% (3 patients) experienced progressive disease. ORR by independent assessment demonstrated similar responses as reported in Table 4. At the highest treated dose of 7 mg/kg of AFM13 in combination with 200 mg pembrolizumab (cohort 3 + extension, n = 24), the ORR was 88% (21 patients) for both assessments; according to the investigator assessment, 42% (10 patients) experienced a CMR and 46% (11 patients) experienced a PMR. Eight percent (2 patients) had NMR and 4% (1 patient) experienced progressive disease. According to the independent assessment, 46% (11 patients) experienced a CMR, 42% (10 patients) had a PMR, and 3 (13%) experienced progressive disease (Table 4). In the subset of patients who were refractory to BV, 85% (11 patients) achieved an objective response and 46% (6 patients) achieved a CMR.
Tumor ORR summary
| CMR, n (%) | PMR, n (%) | NMR, n (%) | PD, n (%) | ORR, n (%) | |
|---|---|---|---|---|---|
| Investigator assessment | |||||
| Cohorts 1 and 2 (n = 6) | 1 (17%) | 3 (50%) | 0 (0%) | 2 (33%) | 4 (67%) |
| Cohort 3 and extension (n = 24) | 10 (42%) | 11 (46%) | 2 (8%) | 1 (4%) | 21 (88%) |
| Safety analysis set (n = 30) | 11 (37%) | 14 (47%) | 2 (7%) | 3 (10%) | 25 (83%) |
| Investigator assessment | |||||
| Cohorts 1 and 2 (n = 5) | 1 (20%) | 2 (40%) | 2 (40%) | 0 (0%) | 3 (60%) |
| Cohort 3 and extension (n = 24) | 11 (46%) | 10 (42%) | 0 (0%) | 3 (13%) | 21 (88%) |
| Safety analysis set (n = 29) | 12 (42%) | 12 (42%) | 2 (7%) | 3 (10%) | 24 (83%) |
| CMR, n (%) | PMR, n (%) | NMR, n (%) | PD, n (%) | ORR, n (%) | |
|---|---|---|---|---|---|
| Investigator assessment | |||||
| Cohorts 1 and 2 (n = 6) | 1 (17%) | 3 (50%) | 0 (0%) | 2 (33%) | 4 (67%) |
| Cohort 3 and extension (n = 24) | 10 (42%) | 11 (46%) | 2 (8%) | 1 (4%) | 21 (88%) |
| Safety analysis set (n = 30) | 11 (37%) | 14 (47%) | 2 (7%) | 3 (10%) | 25 (83%) |
| Investigator assessment | |||||
| Cohorts 1 and 2 (n = 5) | 1 (20%) | 2 (40%) | 2 (40%) | 0 (0%) | 3 (60%) |
| Cohort 3 and extension (n = 24) | 11 (46%) | 10 (42%) | 0 (0%) | 3 (13%) | 21 (88%) |
| Safety analysis set (n = 29) | 12 (42%) | 12 (42%) | 2 (7%) | 3 (10%) | 24 (83%) |
PD, progressive disease.
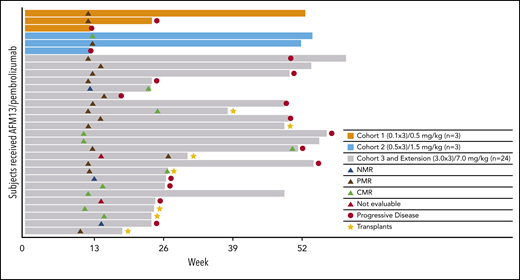
Figure 1 shows a waterfall plot with the respective relative changes in tumor volume during study treatment. The duration and deepening of response are shown in the swimmer’s plot (Figure 2). Of the total patients (N = 30), the vast majority (21 of 25) of the responders (investigator assessment; Table 4) achieved their best response at their first tumor assessment (ie, at ∼13 weeks); 4 patients achieved their best objective response at the ∼26-week assessment or later (Figure 2). A patient’s response was considered to be “deepened” when the patients best overall response improved over time. There was a deepening of response for 1 patient, showing a shift from NMR (no change in 18F fluorodeoxyglucose uptake from baseline) to a CMR (at ∼13 weeks to ∼26 weeks), 2 patients showed a shift from PMR to CMR (2 patients, at ∼13 weeks to ∼26 weeks), whereas 1 patient was not evaluable and shifted to PMR (∼13 weeks to ∼26 weeks) (Figure 2). Responses did not exactly occur at 13, 26, and 52 weeks because response evaluations did not all exactly coincide with the specified time points. The median duration of any response, CMR, and PMR for all patients was 9.9 months (95% confidence interval [CI], 8.4-not estimable [NE]), 10.4 months (95% CI, 2.8-10.4), and 9.0 months (95% CI, 8.4-NE), respectively. The median duration of any response, CMR, and PMR for patients in the MAD cohort was 9.0 months (95% CI, 8.4-10.4), 10.4 months (95% CI, 2.8-10.4), and 8.7 months (95% CI, 8.4-NE), respectively. Duration of response for individual patients is shown in supplemental Figure 2 as percent change from baseline over time. Five patients under study became eligible for stem-cell transplantation. Two additional patients successfully transitioned to SCT while off-study (Figure 2).

Best response according to tumor volume. The dashed line (−30%) represents clinically meaningful responses (30% reduction from baseline). *All assessments are based on CT scan, CRs are based on PET scans (metabolic assessment) and appear opaque on CT scans. NE, not evaluable; PR, partial response; SD, stable disease.
Best response according to tumor volume. The dashed line (−30%) represents clinically meaningful responses (30% reduction from baseline). *All assessments are based on CT scan, CRs are based on PET scans (metabolic assessment) and appear opaque on CT scans. NE, not evaluable; PR, partial response; SD, stable disease.
Discussion
Approximately 10% to 30% of patients with HL relapse after or are refractory to first-line chemotherapy. The standard of care for patients with R/R HL is salvage chemotherapy followed by ASCT, which results in long-term remission for ∼50% of patients.2,3 A milestone was achieved in HL treatment with the development of BV in post-ASCT consolidation or ASCT failure setting.21 BV as monotherapy has an ORR of 75% and CR rate of 34%; however, outside of the consolidation setting, most patients will progress on BV and require further treatment.8,21
The immune system interacts with developing tumors (ie, cancer immunoediting) where tumor cells are initially eliminated by the innate immune system in which the immune system selects tumor variant generation, followed by immune escape where these variants expand in an unchecked manner.22,23 HL tumor cells (Hodgkin and Reed-Sternberg cells) use various mechanisms to immunoevade detection, including downregulation of surface MHC class I and II expression.21 The PD-1/PD-L1 axis is one of the major escape mechanisms in HL and provides strong rationale for immune checkpoint inhibitors as treatment of patients with HL, revolutionizing therapy protocols over the past 5 years.21,24
Both pembrolizumab and nivolumab have been approved for the treatment of R/R HL after ASCT and BV failure, whereas pembrolizumab is also approved for the treatment of transplant-ineligible patients after BV failure. In a phase 2 study, pembrolizumab showed good antitumor activity for patients with R/R HL (3 cohorts: ASCT followed by BV; ASCT, but without BV after transplant; salvage chemotherapy and BV), with an ORR of 69.0% and CR rate of 22.4% at 10.1 months.9 With a follow-up of 27.6 months, ORR was 71.9% and CR of 27.6%, with duration of response at 16.5 months in all patients.25 Pembrolizumab has antitumor activity with acceptable tolerability in patients with R/R HL for those treated with BV before or after ASCT and hard-to-treat chemoresistant patients. However, there remains the unmet need for improved treatment of patients with R/R HL in these treatment groups.
NK cells are cytotoxic lymphocytes of innate immunity and are essential for immunosurveillance of infections and cancer.26 At high antigen density, NK cells can kill target cells opsonized with antibodies by ADCC after recognition of the Fc portion of immunoglobulin (Ig) with specific Fc receptors. At limiting antigen density on target cells, an insufficient degree of opsonization of the target cell by IgG leads to a low level of ADCC and thus tumor immune escape because of few low-affinity interactions between the Fc domain and Fc receptors.15 Interestingly, NK cells are also exhausted by the PD-1/PDL-1 axis in HL and this is reversible with immune checkpoint inhibitors.21,27 CD16A, known as FcγRIIIA receptor, is the main receptor facilitating ADCC via activation of NK cells characterized by release of cytotoxic granules, death receptor signaling, and release of pro-inflammatory cytokines. Following activation, NK cells release preformed granules containing pore-forming perforin and granzymes which trigger apoptosis of target cells.15 In addition to NK cells, CD16A is expressed on macrophages, monocytes, and γ/δ T cells.15
Multiple NK cell-engaging antibodies, as well as bispecific antibodies, targeting immune effector cells and tumor cells, are in development. AFM13 is a tetravalent bispecific antibody that targets CD16A/CD30, derived from the ROCK platform.14,18 The platform is equipped with unique CD16A-specific antibody variable domains optimized for affinity and avidity to achieve sustainable activation of NK cells and macrophages.13 The CD16A antibody variable domain of AFM13 binds to a specific epitope on CD16A distinct from the Fc-binding site, resulting in a high binding affinity that is only slightly inhibited by serum IgG binding. ROCK-antibody innate cell engagers are designed to be complementary to other immunotherapeutic approaches, such as agents that stimulate adaptive immunity and adoptive NK cellular therapies.13
In the phase I AFM13 monotherapy study in heavily pretreated HL patients, in 13 patients who received doses of ≥1.5 mg/kg AFM13, the ORR was 23% and the disease control rate was 77%. AFM13 was found to be well-tolerated, safe (MTD was not reached), and active for heavily pretreated patients with HL, including BV-refractory patients.18 CD30 expression levels are sustained on lymphoma cells from patients with HL that are refractory to, or have relapsed after, treatment with BV, allowing specific targeting of tumor cells.19
The treatment of AFM13 in combination with pembrolizumab was well tolerated, with similar safety profiles compared with the known profiles of each agent alone. IRR and its associated symptoms continued to be the most common AE related to AFM13 (93%), as was observed in a phase 1 study of AFM13 as monotherapy (68%).18 As in the phase 1 study, these events generally resolved with standard treatment measures. Although this AFM13 and pembrolizumab combination trial was a small clinical study, it appears that the incidence of IRR related to AFM13 is somewhat higher when administered in combination with pembrolizumab. A larger randomized study would be required to draw a definitive conclusion on the effect of the combination on the incidence of AFM13-related IRR. Two patients from part 2 (extension study) of the study reported adverse events of clinical interest (AST increased). Both cases corresponded to transient transaminase elevations, with no relevant clinical signs of inflammation, cholestasis, nor additional functional liver parameter increase.
All patients in this study had relapsed after or were refractory to BV treatment. BV targets CD30 but does not diminish or downregulate CD30 in refractory patients.19 Therefore, prior BV treatment would not be expected to preclude use of AFM13 after BV treatment of R/R HL disease. Notably, the subset of patients in this study who were refractory to BV seemed to respond as well to the combination treatment, as did the overall population with 85% (11 patients) achieving an objective response and 46% (6 patients) a CMR.
A limitation of this phase 1b single-arm noncomparative study design is the inability to assess the individual contribution of AFM13 vs pembrolizumab to the efficacy observed. None of the patients had received prior anti-PD1/PD-L1 therapy (ie, they were all pembrolizumab naïve). At the time this clinical trial began (June 2016), anti-PD-1 therapy had not yet been approved for use (US Food and Drug Administration approval for pembrolizumab in patients with R/R HL was granted in March 2017); hence, it would not have been feasible at that time to enroll patients that had undergone prior anti-PD-1 therapy. Nevertheless, the overall response and CMR rates in this study demonstrate early signs of clinical activity.
This phase 1 study data support further investigation into the combination of an innate cell engager with a therapy such as an immune checkpoint inhibitor that stimulates the adaptive immune system. Ongoing or planned clinical trials for AFM13 include a phase 2 trial to optimize the treatment schedule for R/R HL (NCT02321592), AFM13 for R/R CD30+ cutaneous lymphomas (NCT03192202), a combination of cord blood-derived NK cells, and AFM13 for patients with R/R CD30+ lymphoma (NCT04074746). A registration-directed international phase 2 study is also ongoing to assess AFM13 in patients with R/R CD30+ T-cell lymphoma or transformed mycosis fungoides (REDIRECT, NCT04101331).
In summary, treatment with AFM13 in combination with pembrolizumab was well-tolerated with adverse events that were generally manageable with standard treatment and demonstrated clinical and pharmacodynamic activity.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors express gratitude to the patients and their families for participation in the study and thank Dacia Chase and John Facciponte, from the W2O Group, and Jeremy Henriques for writing and editorial services supported by Affimed GmbH.
This study was supported by Affimed GmbH.
Authorship
Contribution: N.L.B., A.F.H., E.D.-D., A.M., A.F.-T., R.G.-S., P.A., S.D., A.R.I., I.S.L., C.R., T.S., R.C., and S.M.A. participated in the study and enrolled and treated patients on the clinical trial; S.E.S., A.S., and K.P. were employees of the Sponsor, Affimed GmBH; L.A. and C.C.-J. were employees of the Sponsor, Affimed Inc.; and all authors reviewed and/or edited all drafts of the manuscript, including the figures and tables.
Conflict-of-interest disclosure: N.L.B. reports grant funding from Affimed, BMS, and Merck; A.F.H. reports grant funding from AstraZeneca, BMS, Gilead, Immune Design, KiTe Pharma, Merck, and Seattle Genetics, and personal fees from Adaptive Biotechnologies, BMS, Genentech, KiTe Pharma, Merck, and Seattle Genetics; E.D.-D. reports personal fees from BMS and Takeda and nonfinancial support from Roche and Takeda; S.M.A. reports grant funding from Affimed, AI Therapeutics, BMS, Regeneron, Seattle Genetics, and Trillium; A.M. reports grant funding from Affimed, ADC Therapeutics, ASTEX, BMS, Celgene, FortySeven Inc., Gilead, Incyte, KiTe Pharma, Juno, Merck, Oncotartis, Pharmacyclics, Rhizen, Roche-Genentech, Seattle Genetics, and Takeda, and personal fees from AstraZeneca, BMS, Celgene, Gilead, KiTe Pharma, Juno, Pharmacyclics, and Seattle Genetics; A.F.-T. reports grant funding by UAB and is a former employee of Seattle Genetics; R.G.-S. reports clinical grant funding from Takeda and University Hospital of Salamanca, CME fees from the Spanish Society of Hematology, personal fees from Beyond Spring, Janssen-Cilag, Roche, and Takeda, and grant funding from Gilead; P.A. reports consultancy services from Affimed, Adaptive, ADC Therapeutics, BMS, Celgene, Merck, Morphosys, and Pfizer, grant funding from Affimed, Adaptive, BMS, Genentech, IGM, Merck, Otsuka, Tensha, and Sigma τ, and personal fees from BMS and Merck; and S.E.S., L.A., A.S., K.P., and C.C.-J. are employees of Affimed. The remaining authors declare no competing financial interests.
Correspondence: Stephen Ansell, Division of Hematology, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: ansell.stephen@mayo.edu.

Comments
Correspondence in reference to previously published manuscripts: “Nancy L. Bartlett, et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2020; 136: 2401-2409.”
In blood advance of 20194 and then haematologica of 20205, we reported encouraging single-center efficacy and safety results of a prospective trial in 26 aged <60 years patients (with similar demographics and baseline characteristics to Bartlett et al, in particular resistant to previous treatment with brentuximab vedotin in 30% of cases) receiving a new salvage regimen named Bv+Bs-21 (consisting of 3-days outpatients i.v. infusions of 1.8 mg/kg of brentuximab vedotin on day 1 of each 3-week cycle combined in sequence to bendamustine supercharge on days 2 and 3 of the treatment cycle at a fixed dose of 120 mg/m2 per day, for a total of 4 courses) for R/R c-HL during the 2013-2020 period in Italy. Ten patients (38%) experienced grade ≥3 treatment-related adverse events (CMV reactivation in 7 cases and neutropenia in 3 cases), all resolved without requiring hospitalization. At post-Bv+Bs-21 evaluation, 100% of patients had deep metabolic responses with Deauville 5-point scale scores ≤3; thereafter, 23 patients (77%) received HSCT. At a median follow-up of 33 months, the progression-free survival was 94% for the entire population.
In conclusion, our clinical data indicate that bendamustine (an old and low-cost cytotoxic agent) used in a new schedule modality characterized by great synergy (i.e., increased dose and post first-in-class anti-CD30 drug conjugate infusion) as salvage regimen has high activity against R/R HRS cells.
References
1. Bartlett NL, Herrera AF, Domingo-Domenech E, et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood 2020; 135: 220-226
2. Connors JM. Hodgkin lymphoma: outsmarting HRS cells. Blood 2020; 135: 2362-2364
3. Cohen JB, Wei L, Maddocks KJ, et al Gemcitabine and bendamustine is a safe and effective salvage regimen for patients with recurrent/refractory Hodgkin lymphoma: Results of a phase 1/2 study. Cancer. 2020 Mar 15;126(6):1235-1242.
4. Picardi M, Della Pepa R, Giordano C, et al. Brentuximab vedotin followed by bendamustine supercharge for refractory or relapsed Hodgkin lymphoma. blood advances 2019; 3: 1546-1552
5. Della Pepa R, Picardi M, Pugliese N, et al. Brentuximab vedotin followed by bendamustine supercharge for refractory or relapsed Hodgkin lymphoma: mature results of a monocentric prospective trial. haematologica 2020; 105: S101-102