

Key Points
CPI203-expanded cord blood cells retain bone marrow repopulating capacity.
CPI203 promotes human megakaryocyte development ex vivo.

Abstract
Although cytokine-mediated expansion of human hematopoietic stem cells (HSCs) can result in high yields of hematopoietic progenitor cells, this generally occurs at the expense of reduced bone marrow HSC repopulating ability, thereby limiting potential therapeutic applications. Because bromodomain-containing proteins (BCPs) have been demonstrated to regulate mouse HSC self-renewal and stemness, we screened small molecules targeting various BCPs as potential agents for ex vivo expansion of human HSCs. Of 10 compounds tested, only the bromodomain and extra-terminal motif inhibitor CPI203 enhanced the expansion of human cord blood HSCs without losing cell viability in vitro. The expanded cells also demonstrated improved engraftment and repopulation in serial transplantation assays. Transcriptomic and functional studies showed that the expansion of long-term repopulating HSCs was accompanied by synchronized expansion and maturation of megakaryocytes consistent with CPI203-mediated reprogramming of cord blood hematopoietic stem and progenitor cells. This approach may therefore prove beneficial for ex vivo gene editing, for enhanced platelet production, and for the improved usage of cord blood for transplantation research and therapy.
Introduction
Gene regulation through epigenetic modification is vital for normal and malignant hematopoiesis,1 and epigenetic regulators have been highlighted as important therapeutic targets. Small molecules that target chromatin-regulating proteins, such as histone deacetylase inhibitors and DNA demethylating agents, overcome the loss-of-stemness by human hematopoietic stem cells (HSCs) that occurs in ex vivo expansion cultures, but it is unclear if all cell types can be generated by reported protocols, especially megakarocytes2-4 . Bromodomain-containing proteins (BCPs), as components of transcription factor complexes and determinants of epigenetic memory,1,5,6 have been implicated in regulating mouse HSC self-renewal and “stemness,”7,8 but are not well-studied in normal human hematopoiesis. By screening small molecules that target various BCPs in umbilical cord blood (UCB) HSCs, we demonstrate that the bromodomain and extra-terminal motif (BET) domain inhibitor CPI203 promotes ex vivo expansion of long-term repopulating human HSCs and megakaryocytes.
Methods
CD133+ human UCB cells (purity >90%) were cultured under optimized conditions for 5 days in stem cell expansion cultures (with stem cell factor [SCF], TPO, and FLT3L as described previously4,9-11 ). Detailed methods are described in supplemental Materials, available on the Blood Web site. Sequencing data have been submitted to the NCBI Gene Expression Omnibus (GSE140813).
Results and discussion
BET inhibitors support ex vivo expansion of phenotypic HSCs
To investigate the role of BCPs in normal hematopoiesis, we tested 10 BCP inhibitors targeting BRD2-4, BRDT, BAZ2A/B, CREBBP, EP300, SMARCA, or PB1 on UCB CD133+ cells (supplemental Figure 1A). Cells were plated in stem cell expansion media with increasing concentrations of these compounds (60 nM to 15 μM) and then analyzed by flow cytometry (supplemental Table 1). Lin−CD34+CD38−CD45RA−CD90+CD49f+ cells were identified phenotypically as HSCs (pHSCs).12 Only BET inhibitors resulted in increased absolute numbers of pHSCs (supplemental Figure 1B), consistent with previous studies demonstrating increased LSK cells/HSCs in mice administered with JQ1.8 Most (JQ1, PFI-1, bromospirone, OXF-BD-02; supplemental Figure 1B) showed their greatest effects at concentrations >5000 nM, which also reduced cell viability. In contrast, with CPI203, the numbers of pHSCs increased significantly, commencing from concentrations as low as 60 nM, with cells remaining viable up to 540 nM (supplemental Figure 1B).
To further assess CPI203, 5000 UCB CD133+ cells/well were cultured in cytokines with 150 nM CPI203 for 5 days (Figure 1A). Although total nucleated cells steadily increased in the vehicle/cytokine control cultures as expected, mainly from to an expansion of immunophenotypic LMPP (Lin−CD34+CD38−CD45RA+) and Lin+ compartments, total nucleated cells expansion was significantly less in the CPI203/cytokines cultures than in vehicle/cytokines (supplemental Figure 2A). Furthermore, numbers of total Lin−CD34+ HSPCs were not altered by addition of CPI203 compared with the vehicle/cytokine control (P = .90 at day 5) and cell viability was unaffected by CPI203 treatment over time (P = .82 at day 5; supplemental Figure 2A). However, the absolute number of pHSCs started to increase significantly by 24 hours after culture initiation with CPI203 and remained significantly higher than the vehicle/cytokine control in the following a 4-day culture (Figure 1B). Similarly, the increase in absolute numbers of immunophenotypic MPP (Lin−CD34+CD38−CD45RA−CD90−) occurred earlier in CPI203 than vehicle-containing cultures (supplemental Figure 2A).
![Cord blood hematopoietic stem/progenitor cell expansion ex vivo by the BET inhibitor CPI203. (A) Flow plots of 5-day expanded UCB CD133+ cells in vehicle/cytokines (top) and CPI203/cytokines (bottom), showing the expression of Lineage (Lin), CD34, CD38, CD45RA, CD90, and CD49f markers. Sequential gating strategy from left to right. (B) Absolute numbers of phenotypically defined (p) Lin−CD34+CD38−CD45RA−CD90+CD49f+ HSCs increased significantly in CPI203/cytokine condition compared with the vehicle/cytokine control (**P ≤ .01; ***P ≤ .005; each well seeded with 5000 cells, n = 4). (C) Absolute number of LTC-IC per well in 5-day CPI203/cytokine vs vehicle/cytokine expansion cultures based on limiting dilution analysis estimation. (n = 4, **P ≤ .01; ***P ≤ .005). (D) Percentage of Annexin V+PI− (apoptotic) and Annexin V+PI+ (dead) cells in cytokine containing medium supplemented with CPI203 or the vehicle control did not differ on day 5 of expansion (n = 3, NS P = .82. (E) Bone marrow cells harvested from mice injected with 500 unexpanded UCB CD133+ cells or progeny of 500 culture initiating UCB CD133+ cells show significantly more human cells in CPI203/cytokine-expanded conditions than unexpanded or vehicle/cytokines conditions (left, primary transplantation; right, secondary transplantation, all replicates shown in supplemental Figures 3-6 and supplemental Tables 2-5). (F) Dot plots show the percentage of hCD45+ human leukocytes in all bone marrow cells from recipient mice in different conditions for (top) primary transplants and (bottom) secondary transplants (n = 3-4; *P ≤ .05; **P ≤ .01, 1-way analysis of variance with multiple comparison [Fisher’s least significant difference]).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/136/21/10.1182_blood.2020005357/1/m_bloodbld2020005357f1.png?Expires=1767698958&Signature=H~sbOXHGHlPpsE~sgQxciY~Bf5V2rgUYuEPD6DHB2D7iXEzGBBGVt2AgHPOGwbJpZZkk6IA3gBRcj7k7VbxA7jINq2aXT540u9O63SHP8e0ORZj-Cj0tmQF96VRZx5fe~QHPgTRxVmEuQjrWZ8Gh~Ay~HzExgUGZt8Gcylxlfpu2XOhCJ2PrMNUxAm0vf~EipiOfhaTOE509Jmi5xUQWEYlS6FAU4C8-yLbW9oz4917igRFp0zUjOTMgDF5LQdeGpp9sl8BkiFe3rzoXW5m7OVWkGYB4-eW5eNhB-Kid9GluJ2rSM6iPsmgcwvt97zi-V50kX7v2ALfmv3oqnOAvhQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Cord blood hematopoietic stem/progenitor cell expansion ex vivo by the BET inhibitor CPI203. (A) Flow plots of 5-day expanded UCB CD133+ cells in vehicle/cytokines (top) and CPI203/cytokines (bottom), showing the expression of Lineage (Lin), CD34, CD38, CD45RA, CD90, and CD49f markers. Sequential gating strategy from left to right. (B) Absolute numbers of phenotypically defined (p) Lin−CD34+CD38−CD45RA−CD90+CD49f+ HSCs increased significantly in CPI203/cytokine condition compared with the vehicle/cytokine control (**P ≤ .01; ***P ≤ .005; each well seeded with 5000 cells, n = 4). (C) Absolute number of LTC-IC per well in 5-day CPI203/cytokine vs vehicle/cytokine expansion cultures based on limiting dilution analysis estimation. (n = 4, **P ≤ .01; ***P ≤ .005). (D) Percentage of Annexin V+PI− (apoptotic) and Annexin V+PI+ (dead) cells in cytokine containing medium supplemented with CPI203 or the vehicle control did not differ on day 5 of expansion (n = 3, NS P = .82. (E) Bone marrow cells harvested from mice injected with 500 unexpanded UCB CD133+ cells or progeny of 500 culture initiating UCB CD133+ cells show significantly more human cells in CPI203/cytokine-expanded conditions than unexpanded or vehicle/cytokines conditions (left, primary transplantation; right, secondary transplantation, all replicates shown in supplemental Figures 3-6 and supplemental Tables 2-5). (F) Dot plots show the percentage of hCD45+ human leukocytes in all bone marrow cells from recipient mice in different conditions for (top) primary transplants and (bottom) secondary transplants (n = 3-4; *P ≤ .05; **P ≤ .01, 1-way analysis of variance with multiple comparison [Fisher’s least significant difference]).
Cord blood hematopoietic stem/progenitor cell expansion ex vivo by the BET inhibitor CPI203. (A) Flow plots of 5-day expanded UCB CD133+ cells in vehicle/cytokines (top) and CPI203/cytokines (bottom), showing the expression of Lineage (Lin), CD34, CD38, CD45RA, CD90, and CD49f markers. Sequential gating strategy from left to right. (B) Absolute numbers of phenotypically defined (p) Lin−CD34+CD38−CD45RA−CD90+CD49f+ HSCs increased significantly in CPI203/cytokine condition compared with the vehicle/cytokine control (**P ≤ .01; ***P ≤ .005; each well seeded with 5000 cells, n = 4). (C) Absolute number of LTC-IC per well in 5-day CPI203/cytokine vs vehicle/cytokine expansion cultures based on limiting dilution analysis estimation. (n = 4, **P ≤ .01; ***P ≤ .005). (D) Percentage of Annexin V+PI− (apoptotic) and Annexin V+PI+ (dead) cells in cytokine containing medium supplemented with CPI203 or the vehicle control did not differ on day 5 of expansion (n = 3, NS P = .82. (E) Bone marrow cells harvested from mice injected with 500 unexpanded UCB CD133+ cells or progeny of 500 culture initiating UCB CD133+ cells show significantly more human cells in CPI203/cytokine-expanded conditions than unexpanded or vehicle/cytokines conditions (left, primary transplantation; right, secondary transplantation, all replicates shown in supplemental Figures 3-6 and supplemental Tables 2-5). (F) Dot plots show the percentage of hCD45+ human leukocytes in all bone marrow cells from recipient mice in different conditions for (top) primary transplants and (bottom) secondary transplants (n = 3-4; *P ≤ .05; **P ≤ .01, 1-way analysis of variance with multiple comparison [Fisher’s least significant difference]).
To estimate the proportion of long-term culture-initiating cells (LTC-IC) in the pHSC subset, we performed a limiting dilution analysis on pHSCs sorted before and after 5 days of expansion by CPI203/cytokines or vehicle/cytokines. LTC-IC frequency was almost identical (1 in 42.4 vs 1 in 42.2 for day 5 cells for day 5 CPI203-treated and vehicle-treated pHSCs, respectively; supplemental Figure 2B). However, because the absolute number of pHSCs in CPI203/cytokine cultures was 5- to 10-fold higher than the vehicle/cytokine control from day 2 of culture (Figure 1B), and cell viability was unaffected by CPI203 (P = .67; Figure 1D; supplemental Figure 2C), we estimate that CPI203 expanded absolute numbers of LTC-ICs 5- to 10-fold compared with a minimal expansion by vehicle/cytokines (Figure 1C; P < .001, n = 4). As expected, although the LTC-IC frequency was higher in unexpanded pHSCs (1 in 12.9), consistent with the pHSC frequency (0.1 ± 0.2% in CD133+ cells) and similar to that observed in previous repopulation studies using irradiated NSG mice,12 the total numbers of LTC-IC in the pHSC fraction of CPI203 cultures were 1.5 to 3 times higher from days 2 to 5 compared with the unexpanded pHSCs.
CPI203/cytokine-expanded CD133+ cells show improved bone marrow engraftment in nonirradiated NOD,B6.SCID Il2rg−/−KitW41/W41 mice
To confirm preservation of HSC function of CPI203-expanded cells in vivo, unexpanded CD133+ cells (500-50 000), or the total progeny of that cell number after expansion, were injected into nonirradiated adult NOD,B6.SCID Il2rg−/−KitW41/W41 mice (supplemental Table 2). After 20 to 22 weeks, all 14 mice injected with CPI203/cytokine-expanded cells engrafted the bone marrow (BM; ≥1% human [h]CD45+ cells) at all cell doses, whereas consistent engraftment of vehicle/cytokine-expanded cells only occurred with higher cell doses (Figure 1E-F; supplemental Tables 2-4). Indeed, the level of BM engraftment in the 500-cell initiating group was 23.1% ± 5.9% in CPI203/cytokine conditions vs 1.5% ± 1.4% for vehicle/cytokines (Figure 1E-F). Multilineage reconstitution was observed, with substantial hCD235a+ erythroid cells (40.1% ± 15.2%), as well as lymphocytes (CD19+ B cells: 20.1% ± 5.9%; CD3+ T cells: 1.4% ± 1.2%), CD33+ myeloid cells (0.7% ± 0.2%), and Lin-CD34+ HSPCs (0.2% ± 0.1%), detected in the total BM of engrafted mice (supplemental Figure 3A-F; supplemental Tables 3 and 4). Spleen and peripheral blood engraftment with hCD45+ cells were also higher for CPI203/cytokine expanded cells in 500-cell initiating group: spleen, 37.6% ± 13.2% vs 4.5% ± 3.3%; peripheral blood, 5.8% ± 3.2% vs 0.2% ± 0.2%) (supplemental Figure 4A-C; all data in other cell dosage appear in supplemental Figure 4 and supplemental Table 4).
To confirm long-term repopulating capacity of CPI203/cytokine-treated cells, secondary transplantation was performed and analyzed 22 weeks after injection. All mice injected with BM cells from mice receiving CPI203/cytokine-expanded cells engrafted (7.2% ± 2.9% hCD45+ cells in BM) with multilineage reconstitution, compared with 0.3% ± 0.2% hCD45+ cells in mice transplanted with the same number of BM cells from mice receiving vehicle/cytokine-expanded cells (Figure 1E-F; supplemental Figures 5 and 6; supplemental Table 5) confirming that CPI203 can expand phenotypic and functional long term (LT)-HSC.
CPI203 promotes expansion of HSC and megakaryocytes
Finally, to investigate the transcriptomic effects of CPI203 on UCB HSPCs, we performed RNA sequencing on cells expanded with CPI203/cytokines or vehicle/cytokines. Differences in expression of many HSC-related genes were detected (Figure 2A; supplemental Figure 7A-B; supplemental Spreadsheets). Genes highly expressed in HSCs (PROM1, POU5F1, EMCN, CRHBP, HLF, MEIS1, JUN, and CXCR42,9,13-16 ) were significantly more highly expressed in CPI203/cytokine-expanded cells, in keeping with the higher numbers of pHSCs. In contrast, myeloid-, erythroid-, and lymphoid-related genes,13 including CSF3R, PRTN3, CA1, CNRIP1, APOC1, TFR2, CD4, CD79A, and CD79B, were more highly expressed in vehicle/cytokine conditions. Unexpectedly, 3 of the most upregulated genes in CPI203/cytokine-expanded cells are strongly associated with megakaryocyte (MK) development (CXCR4,17 PF4,18 and C6orf2519 ).
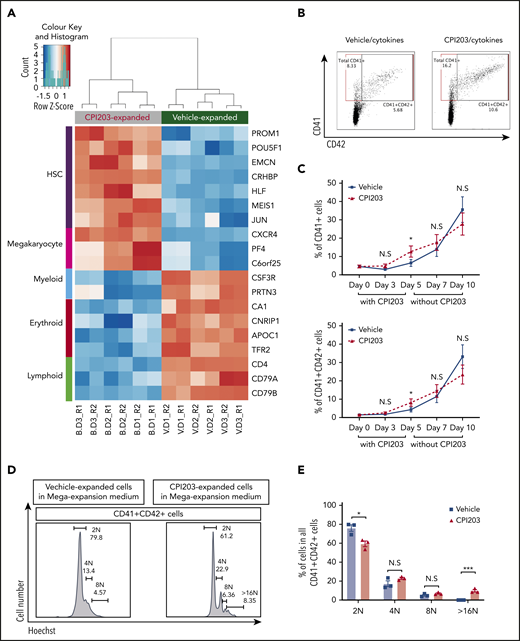
CPI203 promotes megakaryocyte-accompanied ex vivo expansion of CB HSCs. (A) RNA sequencing was performed on UCB CD133+ cells that had been expanded for 5 days in culture with CPI203/cytokines (B) or vehicle/cytokines (V). Top differentially expressed genes associated with HSCs, megakaryocytes, myeloid cells, erythroid cells, and lymphoid cells are shown in a heatmap (false discovery rate < 0.01, gene expression level from low to high shown as blue to orange). (B) Flow plots showing the total CD41+ or CD41+CD42+ cells detected in all cells expanded in CPI203/cytokines condition compared with vehicle/cytokines on day 5. (C) Increased total CD41+ (top) and CD41+CD42+ (bottom) megakaryocytic cells expanded in CPI203/cytokine conditions compared with vehicle/cytokine for 5 days, followed by a further 5 days’ expansion in MK expansion medium (TPO, SCF) without CPI203 or vehicle (n = 3; *P ≤ .05). (D-E) Increased ploidy of megakaryocytes generated in MK expansion medium from d5-CPI203/cytokine-expanded cultures compared with d5-vehicle/cytokine-expanded cultures (n = 3; *P ≤ .05; ***P ≤ .005). Data shown are from day 10 of extended cultures: 5 days in CPI203/cytokines or vehicle/cytokines followed by 5 days in MK expansion medium (SCF, TPO) without CPI203 or vehicle.
CPI203 promotes megakaryocyte-accompanied ex vivo expansion of CB HSCs. (A) RNA sequencing was performed on UCB CD133+ cells that had been expanded for 5 days in culture with CPI203/cytokines (B) or vehicle/cytokines (V). Top differentially expressed genes associated with HSCs, megakaryocytes, myeloid cells, erythroid cells, and lymphoid cells are shown in a heatmap (false discovery rate < 0.01, gene expression level from low to high shown as blue to orange). (B) Flow plots showing the total CD41+ or CD41+CD42+ cells detected in all cells expanded in CPI203/cytokines condition compared with vehicle/cytokines on day 5. (C) Increased total CD41+ (top) and CD41+CD42+ (bottom) megakaryocytic cells expanded in CPI203/cytokine conditions compared with vehicle/cytokine for 5 days, followed by a further 5 days’ expansion in MK expansion medium (TPO, SCF) without CPI203 or vehicle (n = 3; *P ≤ .05). (D-E) Increased ploidy of megakaryocytes generated in MK expansion medium from d5-CPI203/cytokine-expanded cultures compared with d5-vehicle/cytokine-expanded cultures (n = 3; *P ≤ .05; ***P ≤ .005). Data shown are from day 10 of extended cultures: 5 days in CPI203/cytokines or vehicle/cytokines followed by 5 days in MK expansion medium (SCF, TPO) without CPI203 or vehicle.
We therefore examined the percentage of MK in the cultures. After expansion in CPI203/cytokines for 5 days (Figure 2B-C), the proportion of both CD41+ and CD41+CD42+ MK was significantly (∼2-fold) higher than for the vehicle/cytokine control. To further test the effect of CPI203 on MK differentiation and maturation, the day 5 CPI203/cytokines or vehicle/cytokines expanded cells were transferred into MK-supportive media (SCF, TPO) and cultured without CPI203 or vehicle for a further 5 days. The percentages of CD41+ and CD41+CD42+ MK increased in the second culture phase (Figure 2C), and, by day 10, MK derived from the d5-CPI203-expanded cells were of higher ploidy than the d5-vehicle/cytokine-expanded UCB HSPCs (Figure 2D-E). This suggests that CPI203 promotes MK maturation, and is consistent with previous work showing that BET inhibition promotes enlargement, polyploidization and proplatelet formation of human UCB MK-progenitors.20 Whether BET inhibition might also exert effects through reprogramming toward MK-biased HSCs is an interesting and pertinent question, but further work would be needed to address this.
In summary, we demonstrate that the BET inhibitor CPI203 expands serially transplantable LT-HSCs ex vivo and, in contrast to UM171, which inhibits erythroid and megakaryocytic differentiation during ex vivo expansion of LT-HSCs,21 CPI203 also supports megakaryocyte maturation. CPI203 may therefore prove beneficial for enhanced platelet production and for the improved usage of UCB for transplantation. Understanding the mechanisms by which CPI203 exerts these effects should provide fundamental insight into MK development.
The sequencing data reported in this article have been submitted to the NCBI Gene Expression Omnibus database (accession number GSE140813).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
The online version of this article contains a data supplement.
Acknowledgments
These studies received grant support from an MRC Discovery Award led by Professor Doug Higgs (P.H., J.H., G.A., A.R., J.D., T.A.M., I.R., F.I., S.M.W.; reference MC_PC_15069). J.D. and P.H. are also funded by an MRC Clinician Scientist Award (Fellowship reference MR/R008108) to J.D. J.H. is a recipient of KRUK Senior Fellowship, G.A. is a recipient of a Clarendon Scholarship, F.I. and A.R. are both recipients of Wellcome Trust CRCD Fellowships. R.L. is funded by a Bloodwise project grant and B.P. is a recipient of Cancer Research UK Advanced Clinician Scientist Fellowship. T.A.M. was funded by Medical Research Council Molecular Haematology Unit grant MC_UU_12009/6 and is supported by the NIHR Oxford Biomedical Research Centre Haematology Theme.
Authorship
Contribution: P.H. conceived, designed, performed, analyzed experiments, performed bioinformatics analysis, and wrote the manuscript; J.H., G.A., and F.I. performed in vivo experiments; R.L. and B.P. performed the megakaryocyte analysis; T.J. contributed to the bioinformatics analysis; A.R., C.J.R.B., and G.M.W. supplied chemicals and contributed to experimental analysis; S.M.W., F.I., A.J.R., and T.A.M. designed and supervised the project; S.M.W., F.I., A.J.R., T.A.M., A.R., I.R., J.D., and B.P. analyzed experiments and contributed to writing the manuscript; and all authors reviewed and agreed the final submitted manuscript.
Conflict-of-interest disclosure: J.D. is a cofounder of Nucleome Therapeutics Ltd., to which he provides consultancy. G.M.W. is an employee and minor shareholder (<5%) of OxStem Ltd. T.A.M. is a founding and minor (<5%) shareholder of OxStem Oncology, a subsidiary company of OxStem Ltd. A.J.R. is a founding and minor (<5%) shareholder of OxStem Ltd. The remaining authors declare no competing financial interests.
Correspondence: Peng Hua, Weatherall Institute of Molecular Medicine/Nuffield Division of Clinical Laboratory Medicine, Radcliffe Department of Medicine, John Radcliffe Hospital, Oxford, OX3 9DS, United Kingdom; e-mail: peng.hua@ndcls.ox.ac.uk; and Suzanne M. Watt, Weatherall Institute of Molecular Medicine/Nuffield Division of Clinical Laboratory Medicine, Radcliffe Department of Medicine, John Radcliffe Hospital, Oxford, OX3 9DS, United Kingdom; e-mail: suzanne.watt@ndcls.ox.ac.uk.
