Abstract
Affinity maturation and terminal differentiation of B cells via the germinal center reaction is a complex multistep process controlled by transcription factors that induce or suppress large dynamic transcriptional programs. This occurs via the recruitment of coactivator or corepressor complexes that epigenetically regulate gene expression by post-translationally modifying histones and/or remodeling chromatin structure. B-cell–intrinsic developmental programs both regulate and respond to interactions with other cells in the germinal center that provide survival and differentiation signals, such as T-follicular helper cells and follicular dendritic cells. Epigenetic and transcriptional programs that naturally occur during B-cell development are hijacked in B-cell lymphoma by genetic alterations that directly or indirectly change the function of transcription factors and/or chromatin-modifying genes. These in turn skew differentiation toward the tumor cell of origin and alter interactions between lymphoma B cells and other cells within the microenvironment. Understanding the mechanisms by which genetic alterations perturb epigenetic and transcriptional programs regulating B-cell development and immune interactions may identify opportunities to target these programs using epigenetic-modifying agents. Here, we discuss recently published studies centered on follicular lymphoma and diffuse large B-cell lymphoma within the context of prior knowledge, and we highlight how these insights have informed potential avenues for rational therapeutic interventions.
Introduction
Translocations of the MYC and BCL6 genes are long-standing examples of how genetic alterations in lymphoma can perturb important transcriptional programs. More recently, studies have revealed additional transcription factors, corepressor/coactivator complex components, histone-modifying enzymes, chromatin-remodeling complex components, and chromatin structural components that are targeted by genetic alterations in B-cell lymphoma. These alterations differ in frequency between histologies1 and/or within transcriptionally or genetically defined subtypes,2,3 function by perturbing epigenetic and transcriptional programs that control cellular pathways and cell fate decisions that are important for the tumor’s cell of origin (reviewed elsewhere4-6 ), and are potentially targetable by an increasing number of epigenetic-modifying agents.7 Therefore, understanding the epigenetic basis for lymphoma is an important challenge due to the high potential for clinical translation that could improve patient outcomes. Here, we discuss recently published (2018–2020) studies8-14 and their translational implications.
EZH2: from H3K27me3 to immune synapse disruption
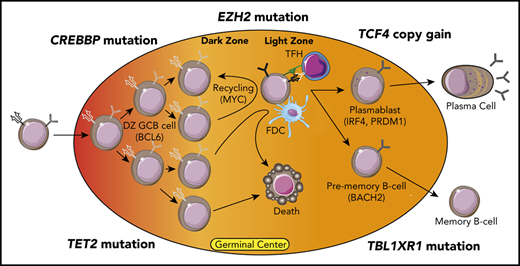
The EZH2 gene encodes a lysine methyltransferase enzyme that catalyzes trimethylation of H3K27 (H3K27me3) as part of the polycomb repressor (PRC) 2 complex. EZH2 is highly expressed in germinal center B (GCB) cells15-17 and normally functions to repress the expression of genes highly expressed in naïve B cells. Gene silencing also occurs in cooperation with BCL6 and BCOR18 to temporarily repress the expression of transcription factors involved in plasma cell differentiation, such as PRDM1, IRF4, and XBP1,19,20 and negative regulators of the cell cycle, such as CDKN1A/B.19 Germinal center (GC)-specific conditional knockout (cKO) of Ezh2 in transgenic mouse models prevents GC development,19 but this can be rescued by co-deletion of the BCL6 target gene, Cdkn1a, highlighting an important role for Ezh2 in controlling GCB cell proliferation.21
Mutations of EZH2 occur in 15% to 25% of follicular lymphoma (FL) and 5% to 10% of diffuse large B-cell lymphoma (DLBCL)4 (25% to 45% of the C3/EZB subtype of DLBCL2,3 ; EZH2 mutation is a seed feature for the EZB subtype). These mutations most often encode a single–amino acid change at Y641 in the catalytic SET domain, causing a neomorphic change in activity that results in higher levels of H3K27me3.22,23 Murine models using viral transduction or conditional knock-in showed that the expression of Ezh2-Y641 mutants results in GC hyperplasia after immunization and cooperates with Bcl2 overexpression to drive lymphomagenesis.19,20 Recent studies have shown that this may result from deepening repression of canonical Ezh2 target genes within GCB cells, as well as spreading of the H3K27me3 mark to a large number of neighboring promoters8 (de novo targets). This spreading may be restricted by 3-dimensional chromatin architecture, leading to gain of H3K27me3 within topologically associated domains and coordinated repression of neighboring tumor suppressor genes.9 By performing targeted sequencing of 57 genes and immunohistochemistry for major histocompatibility complex (MHC) class I and class II on a cohort of DLBCL tumors, Ennishi et al10 identified an association between EZH2 mutations and dual loss of MHC class I and class II or single loss of MHC class I. GCB-like DLBCL tumors with loss of MHC expression also tended to have reduced frequencies of tumor-infiltrating CD4 and CD8 T cells,10 but a subsequent comparison of DLBCL tumors by EZH2 mutation status alone observed no significant differences between EZH2 wild-type and mutant cases.8
Despite a prominent role for EZH2 in regulating BCL6 target gene expression within dark zone (DZ) GCB cells,18,21 Béguelin et al8 recently observed that Ezh2-Y641F conditional knock-in mice have an accumulation of light zone (LZ) GCB cells and no impediment to terminal differentiation. This was driven by Ezh2-mediated repression of genes involved in immune synapse formation with T-follicular helper (TFH) cells, resulting in a loss of interaction with TFH cells and reduced CD40/CD40LG signaling. This attenuated MYC expression and recycling to the DZ was associated with reduced somatic hypermutation and clonal diversity. Importantly, Ezh2 mutant, but not Ezh2 wild-type, GCB cells were able to maintain GCs in the absence of TFH interaction and CD40 signaling due to their increased association with follicular dendritic cells (FDCs). Ezh2-Y641F murine GCB cells were intermeshed with FDCs within the LZ, and human FL tumors with EZH2 mutations more frequently had dense FDC networks within their follicles. Moreover, Ezh2-Y641F murine GCB cells were sensitive to FDC blockade. EZH2 mutations may therefore function by uncoupling GCB cells from the normal process of clonal selection, allowing them to survive and proliferate within the LZ independent of their antigen affinity but dependent on survival signals from FDCs (Figure 1).
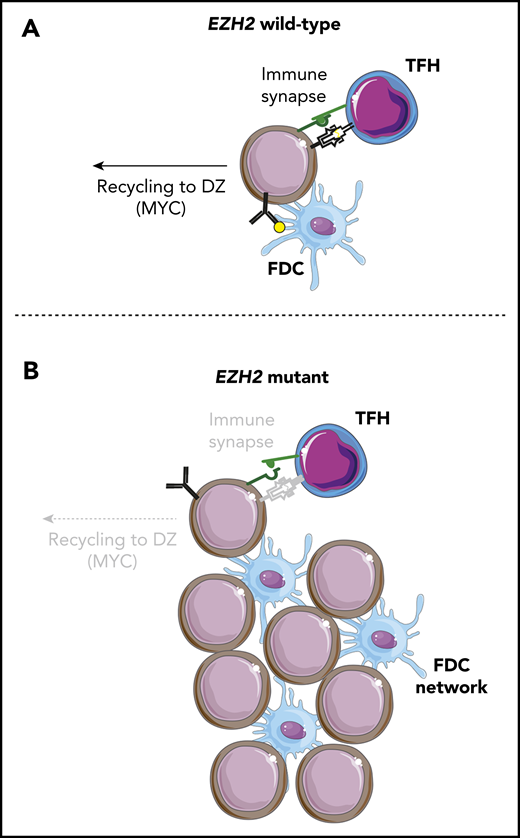
Loss of TFH immune synapse formation and gain of FDC interactions with EZH2 mutations. (A) EZH2 wild-type B cells undergo normal clonal selection by binding antigen on FDCs and presenting it on MHC class II. Those with the highest antigen affinity and presentation form an immune synapse with TFH cells, leading to CD40/CD40L signaling, which stimulates terminal differentiation or DZ recycling. (B) EZH2 mutant B cells have reduced MHC expression and immune synapse formation with TFH cells that leads to decreased CD40/CD40L signaling and DZ recycling. However, these cells are able to proliferate and survive through interactions with an expanded network of FDCs.
Loss of TFH immune synapse formation and gain of FDC interactions with EZH2 mutations. (A) EZH2 wild-type B cells undergo normal clonal selection by binding antigen on FDCs and presenting it on MHC class II. Those with the highest antigen affinity and presentation form an immune synapse with TFH cells, leading to CD40/CD40L signaling, which stimulates terminal differentiation or DZ recycling. (B) EZH2 mutant B cells have reduced MHC expression and immune synapse formation with TFH cells that leads to decreased CD40/CD40L signaling and DZ recycling. However, these cells are able to proliferate and survive through interactions with an expanded network of FDCs.
EZH2 mutant DLBCL is sensitive to catalytic inhibitors of EZH2, providing a rationale for targeting EZH2 in FL and DLBCL. Interim results of the phase 2 study of tazemetostat in relapsed/refractory FL were reported at the 2019 American Society of Hematology annual meeting24 and showed a high response rate in EZH2 mutant tumors. However, responses were also observed in EZH2 wild-type cases. On the basis of the respective response rates and the frequency of EZH2 mutations in FL, if an unselected patient population were treated with tazemetostat, the responders would be expected to consist of approximately equal proportions of EZH2 wild-type and EZH2 mutant cases. Thus, response to EZH2 inhibitors is dictated by more than just EZH2 mutation status. Indeed, EZH2 function is critical for GCB cells, regardless of their mutation status,19 so a subset of EZH2 wild-type tumors may nonetheless be addicted to EZH2 activity. Furthermore, EZH2 suppresses the expression of genes involved in immune synapse formation, such as MHC classes I and II, the expression of which can be promoted on DLBCL cell lines and Ezh2-Y641F murine GCB cells by EZH2 inhibitors.10 Therefore, EZH2 inhibition may also have an immune-potentiating effect. Tazemetostat recently received US Food and Drug Administration approval for treatment of patients with relapsed/refractory FL in whom 2 prior lines of therapy have failed if they have (1) an EZH2 mutation or (2) no satisfactory alternative treatment options. Detailed analysis of the molecular and immunological bases for response/resistance to EZH2 inhibitors, and the redundant or compensatory role for EZH125 in this context, will be required to fully grasp the underlying mechanisms and inform potential combination strategies that may improve efficacy and durability.
CREBBP: not all mutations are created equal
The CREBBP gene encodes a lysine acetyltransferase (KAT) protein that activates gene expression through acetylation of histone H3 lysine 18 (H3K18Ac), histone H3 lysine 27 (H3K27Ac), and other residues. CREBBP is mutated in ∼65% of FL and 10% to 15% of DLBCL4,26 (53% of C3/EZB subtype2,3 ). The majority of mutations encode single–amino acid changes within the catalytic KAT domain that reduce acetyltransferase activity,26 but nonsense/frameshift mutations also occur and are significantly more frequent in DLBCL than in FL.27 CREBBP mutations are associated with poor outcome in FL,28 but KAT domain mutations are associated with a worse progression-free survival than nonsense/frameshift mutations.11 Murine studies using Crebbp knockout (KO)/knockdown found that Crebbp loss promotes B-cell lymphoma in cooperation with Bcl2 overexpression27,29,30 and that regions of reduced histone acetylation associated with Crebbp loss were primarily located at enhancer elements bound by BCL6.29 Consistent with this, cKO of Crebbp in GCB cells leads to reduced expression of genes that are expressed in LZ GCB cells, such as those involved in antigen presentation on MHC class II, B-cell receptor signaling, and interferon signaling.12,29 This is consistent with observations in CREBBP mutant primary tumors, which showed a marked downregulation of MHC class II.31 Importantly, a similar molecular phenotype was observed with cKO of Tet2 in GCB cells due to hydroxymethylation loss that impaired enhancer acetylation,32 suggesting that TET2 mutations (a seed feature of the ST2 DLBCL subtype3 ) and CREBBP mutations may be alternative mechanisms for promoting a BCL6-associated DZ phenotype.
Key differences exist between the magnitudes of changes observed in mouse models using KO/knockdown versus primary tumors in which KAT domain mutations predominate31 and when comparing patient outcomes by CREBBP mutational subtypes. Mondello et al11 therefore investigated functional differences between CREBBP KAT domain mutations and nonsense/frameshift mutations. This was achieved using CRISPR/Cas9 gene editing to generate isogenic lymphoma cell lines with either wild-type CREBBP, CREBBP-R1446C mutation (a mutation hotspot in the KAT domain), or homozygous frameshift mutation (KO). The R1446C mutants had a more severe loss of H3K27Ac than KO mutants, which was linked with a greater reduction in expression of genes with a role in antigen presentation and interferon signaling. CREBBP KAT domain mutations may therefore have a dominant-repressive function which exceeds that of nonsense/frameshift mutations. Regions with reduced H3K27Ac were enriched for BCL6 target genes and were bound by both CREBBP and BCL6 in normal GCB cells. BCL6 suppresses gene expression by recruiting corepressors such as the SMRT–HDAC3 complex, suggesting that CREBBP mutation results in an imbalance in the antagonistic activity between CREBBP-mediated acetylation and BCL6/HDAC3-mediated deacetylation.11,29 Consistent with this, HDAC3 inhibition promoted H3K27Ac and expression of genes that were reduced by CREBBP mutation. However, HDAC3 inhibition was capable of inhibiting the growth of both CREBBP wild-type and mutant DLBCL cell lines and patient-derived xenograft models via induction of the BCL6 target gene, CDKN1A. Furthermore, signatures of antigen presentation and interferon signaling were also induced by HDAC3 inhibition in both CREBBP wild-type and mutant cells. Therefore, CREBBP mutation determines the baseline level of antigen presentation and interferon signaling rather than being a prerequisite for its inducibility by HDAC3 inhibition, because of the conserved role of the CREBBP/BCL6–HDAC3 regulatory axis in both contexts11 (Figure 2). Notably, PD-L1 (CD274), a prominent interferon-inducible gene that blunts antitumor T-cell responses, was induced on tumor B cells by HDAC3 inhibition. This prompted investigation of the efficacy of combination HDAC3 inhibitor and PD-L1 blockade using a syngeneic Bcl6-driven lymphoma model. Treatment with HDAC3 inhibitor alone increased tumor-infiltrating Cd4 and Cd8 T cells to an extent similar to that of PD-L1 blockade alone, but their combination synergistically increased tumor-infiltrating T cells and reduced tumor B cells. This suggests that epigenetic modulation of immune response with HDAC3 inhibitors (or EZH2 inhibitors) may be best used in combination with PD1/PD-L1 blockade to prevent interferon-induced adaptive immune suppression.
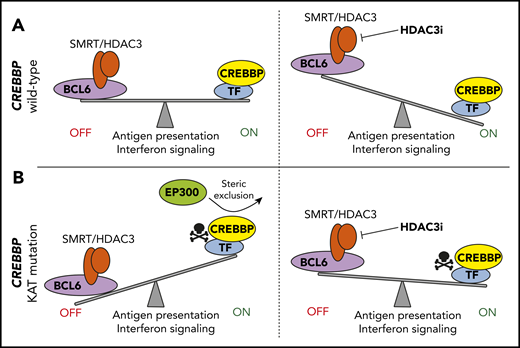
Control of antigen presentation and interferon signaling by CREBBP-mediated antagonism of BCL6/HDAC3. (A) In CREBBP wild-type GCB cells, BCL6 regulates the DZ signature by recruiting the corepressor complexes, including SMRT–HDAC3, to repress its target genes. These genes are reactivated in the LZ by CREBBP. Inhibition of HDAC3 in CREBBP wild-type B cells leads to increased expression of these genes, including those with a role in antigen presentation and interferon signaling, due to the conserved role of the CREBBP/BCL6–HDAC3 regulatory axis in wild-type cells. (B) KAT domain mutation of CREBBP inhibit its catalytic activity and leads to a dominant-repressive effect by preventing the participation of redundant acetyltransferases in transactivation complexes. This leads to loss of antagonism to BCL6-mediated gene repression and reduced expression of antigen presentation and interferon signaling genes. These genes can be restored in CREBBP mutant cells by using an HDAC3-selective inhibitor.
Control of antigen presentation and interferon signaling by CREBBP-mediated antagonism of BCL6/HDAC3. (A) In CREBBP wild-type GCB cells, BCL6 regulates the DZ signature by recruiting the corepressor complexes, including SMRT–HDAC3, to repress its target genes. These genes are reactivated in the LZ by CREBBP. Inhibition of HDAC3 in CREBBP wild-type B cells leads to increased expression of these genes, including those with a role in antigen presentation and interferon signaling, due to the conserved role of the CREBBP/BCL6–HDAC3 regulatory axis in wild-type cells. (B) KAT domain mutation of CREBBP inhibit its catalytic activity and leads to a dominant-repressive effect by preventing the participation of redundant acetyltransferases in transactivation complexes. This leads to loss of antagonism to BCL6-mediated gene repression and reduced expression of antigen presentation and interferon signaling genes. These genes can be restored in CREBBP mutant cells by using an HDAC3-selective inhibitor.
The functional difference between CREBBP-R1446C and KO mutations is consistent with the catalytically inactive mutant CREBBP protein participating in transcriptional coactivating complexes and thereby preventing the participation of redundant acetyltransferases such as EP300.11 A recent study by Meyer et al12 showed, using Crebbp- and Ep300-cKO mice, that, despite having largely nonoverlapping functions with Crebbp in GCB cells, Ep300 becomes critical for GCB cell proliferation in the absence of Crebbp. This was also confirmed in CREBBP mutant DLBCL cell lines using inducible Cas9 KO of EP300 and through the use of CREBBP/EP300 inhibitors. Together, this suggests that paralogous lethality to EP300 inhibition may be an alternative therapeutic approach for CREBBP mutant DLBCL.
C5/MCD subtype of DLBCL: getting in on the action
The C5/MCD genetic subtype of DLBCL is a subset of the activated B-cell (ABC)-like transcriptional subtype, with a propensity for extranodal sites of involvement. The defining genetic characteristics of the C5/MCD subtype are mutations of MYD88 and CD79B that drive chronically active B-cell receptor signaling. DNA copy number gains of chromosome 18q are also found in 48% to 73% of the C5/MCD subtype of DLBCL and have been attributed to the BCL2 oncogene. However, through the analysis of 1000 DLBCL tumors, Jain et al13 recently identified the TCF4 (also called E2-2) transcription factor gene as a more significant and frequent target of 18q gain13 . The immunoglobulin heavy chain (IgM) and MYC were identified as important targets that have enhancers bound by TCF4, higher expression in primary ABC-like DLBCL tumors with TCF4 copy number gains, and increased expression after tetracycline-inducible expression of TCF4 in cell lines. Genetic inhibition of TCF4 function was lethal to ABC-like DLBCL cell lines with TCF4 DNA copy number gains, highlighting it as an attractive therapeutic target. Importantly, the TCF4 gene is one of the most highly BRD4-loaded genes in DLBCL,33 suggesting that BET inhibition may reduce its expression. Prior studies have also shown that ABC-like DLBCL is sensitive to BET inhibition,34 but the underlying mechanism was not clear. Using a BET protein degrader, TCF4 expression was eliminated in DLBCL cell lines with TCF4 copy gain, thereby reducing the expression of IgM and MYC and inducing cell death. MYC is also a direct target of BRD4 and can be directly reduced by BET inhibition.35 However, both cell death and the expression of MYC and IgM were rescued by enforced expression of TCF4 during BET degrader treatment, showing that these phenotypes are at least in part a direct consequence of TCF4 reduction by the BET degrader. Therefore, DNA copy number gains of TCF4 provide a direct mechanistic rationale for the use of BET inhibitors/degraders in the C5/MCD subtype of DLBCL (Figure 3).
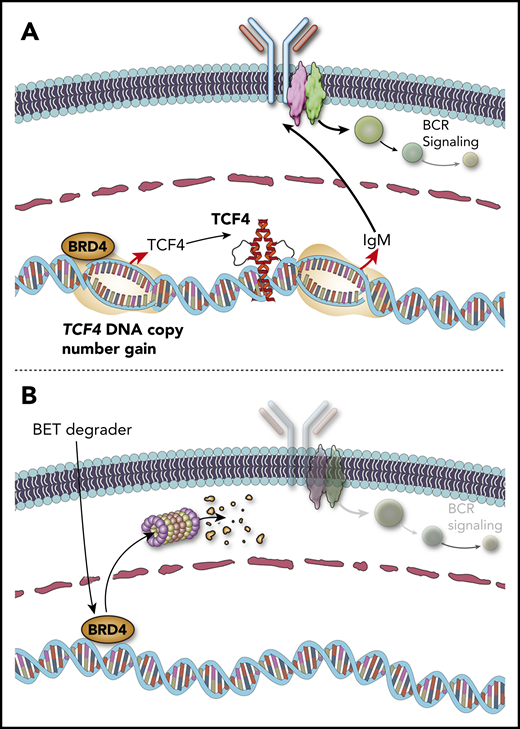
TCF4 DNA copy number gains drive immunoglobulin expression and can be targeted by BET degraders. (A) DNA copy number gains of chromosome 18q increase the expression of the TCF4 (E2-2) transcription factor, which drives increased expression of IgM. (B) The TCF4 gene is regulated by BRD4. BET protein degraders such as ARV-771 eliminate BRD4 protein and reduce the expression of TCF4 and its target genes, including IgM and MYC.
TCF4 DNA copy number gains drive immunoglobulin expression and can be targeted by BET degraders. (A) DNA copy number gains of chromosome 18q increase the expression of the TCF4 (E2-2) transcription factor, which drives increased expression of IgM. (B) The TCF4 gene is regulated by BRD4. BET protein degraders such as ARV-771 eliminate BRD4 protein and reduce the expression of TCF4 and its target genes, including IgM and MYC.
The TBL1XR1 gene encodes a core component of the SMRT–NCOR complex, and mutations of this gene are found in 20% to 35% of the C5/MCD subtype of DLBCL. The mutations primarily alter amino acids on the surface of the WD40 barrel structure that are predicted to have a role in protein–protein interactions. Venturutti et al14 recently modeled these mutations using Tbl1xr1-cKO mice and mice with conditional knock-in of a Tbl1xr1 D370Y mutant allele (Tbl1xr1-D370Y). Using a GCB-specific Cre allele, homozygous cKO and heterozygous Tbl1xr1-D370Y knock-ins were found to have significantly reduced frequencies of GCB cells and smaller GCs after immunization. This is in contrast to most murine lymphoma alleles, which increase or maintain the frequency of GCB cells. Furthermore, this phenotype was not evident with heterozygous cKO mice, suggesting a dominant-negative function for the Tbl1xr1-D370Y mutant allele. The Tbl1xr1-D370Y GCB cells had gene expression patterns reminiscent of human ABC-like DLBCL, with the upregulation of the ABC-like transcriptional signature and derepression of genes with BCL6-regulated enhancers and evidence of expansion of a pre–memory B-cell population. Further analysis revealed that reduced GCB cell frequencies in Tbl1xr1 mutant mice were a consequence of cell cycle arrest of GCB cells and increased rates of GC exit into the memory B-cell compartment. The Tbl1xr1 mutant memory B cells had a reduced frequency of class switching, leading to an IgM-positive bias, which persisted in the periphery over time, and had a significantly higher rate of GC reentry after antigenic rechallenge than wild-type B cells. When crossed with a Bcl2 allele and serially challenged with antigen, Tbl1xr1-cKO mice develop premalignant lesions within extranodal sites, an increased rate of tumor development, and a more immunoblastic appearance compared with mice with the Bcl2 allele alone. Mechanistically, the skew toward a memory B-cell phenotype resulted from a loss of interaction between mutant TBL1XR1 and BCL6 and a concomitant gain of interaction with the BACH2 transcription factor. This resulted in a preferential association of the SMRT–HDAC3 complex with BACH2, an important regulator of memory B-cell development that recruits the SMRT–HDAC3 complex to silence genes involved in plasma cell differentiation, such as PRDM136 (Figure 4). The dependence of this process on the SMRT–HDAC3 complex strongly suggests that HDAC3-selective inhibitors may have a potential role in TBL1XR1 mutant DLBCL.
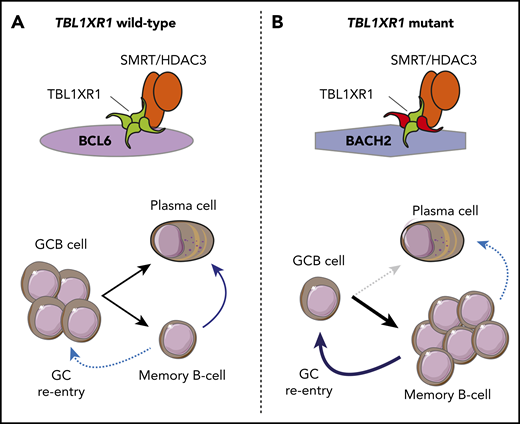
TBL1XR1 mutation promotes a memory B-cell fate and germinal center reentry. (A) In TBL1XR1 wild-type GCB cells, the SMRT–HDAC3 complex preferentially associates with BCL6 and facilitates expansion of GCB cells in the DZ of the germinal center. These can terminally differentiate via the LZ into either plasma cells or memory B cells. Upon antigen rechallenge (blue arrows), memory B cells become antibody-secreting plasma cells, and a small subset reenters germinal centers. (B) Mutant TBL1XR1 (red) acts as a dominant negative to increase association between the SMRT–HDAC3 complex and the BACH2 transcription factor and to decrease association with BCL6. This leads to reduced frequencies of GCB cells and promotes a memory B-cell fate. Upon antigen rechallenge (blue arrows), a reduced frequency of TBL1XR1 mutant memory B cells become plasma cells, and an increased frequency reenter germinal center reactions and undergo additional somatic hypermutation.
TBL1XR1 mutation promotes a memory B-cell fate and germinal center reentry. (A) In TBL1XR1 wild-type GCB cells, the SMRT–HDAC3 complex preferentially associates with BCL6 and facilitates expansion of GCB cells in the DZ of the germinal center. These can terminally differentiate via the LZ into either plasma cells or memory B cells. Upon antigen rechallenge (blue arrows), memory B cells become antibody-secreting plasma cells, and a small subset reenters germinal centers. (B) Mutant TBL1XR1 (red) acts as a dominant negative to increase association between the SMRT–HDAC3 complex and the BACH2 transcription factor and to decrease association with BCL6. This leads to reduced frequencies of GCB cells and promotes a memory B-cell fate. Upon antigen rechallenge (blue arrows), a reduced frequency of TBL1XR1 mutant memory B cells become plasma cells, and an increased frequency reenter germinal center reactions and undergo additional somatic hypermutation.
Concluding remarks
There are still a large number of genetically altered transcriptional and epigenetic regulators that remain to be functionally explored, further details to be discovered regarding the function of previously investigated genes, and an additional level of complexity when considering combinations of genetic alterations that co-occur within the same tumor. Furthermore, epigenetic programs that are important for B-cell development and survival are controlled by epigenetic regulators, signaling pathways, and metabolic programs that are not directly targeted by genetic alterations but could nonetheless serve as therapeutic targets. Therefore, understanding how best to take the increasing yield of epigenetic-modifying agents7,37 and rationally deploy them in a prioritized fashion will require a detailed, descriptive, and functional characterization of the epigenomic architecture of B-cell lymphoma in a manner similar to the approach taken for the coding genome.
Acknowledgments
H.Y. is a fellow of the Leukemia and Lymphoma Society. M.R.G. is a scholar of the Leukemia and Lymphoma Society. Research in M.R.G.’s group is supported by National Institutes of Health grant R01CA201380, the Jaime Erin Follicular Lymphoma Research Consortium, the Futcher Foundation, and the Schweitzer Family Foundation.
Authorship
Contribution: H.Y. and M.R.G. wrote the manuscript.
Conflict-of-interest disclosure: M.R.G. is a consultant for VeraStem Oncology and has stock ownership interest in KDAc Therapeutics. Off-label drug use: None disclosed.
Correspondence: Michael R. Green, Department of Lymphoma/Myeloma, University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 0903, Houston, TX 77030; e-mail: mgreen5@mdanderson.org.