

In this issue of Blood, comprehensively analyze the immunologic and genetic features of a large cohort of adults with hemophagocytic lymphohistiocytosis (HLH).1 The investigators find that natural killer (NK) cells from adults with HLH exhibit an activated phenotype and normal cytotoxic capacity. In contrast, NK cell numbers and interferon-γ (IFN-γ) production are greatly diminished. Genetic studies reveal that 50% of patients harbor ≥1 germline variant of uncertain significance (VUS) in an HLH-associated gene, but none harbor a pathogenic variant.
Immunologic and genetic differences between pediatric and adult HLH. nL, normal; ↑, increased; ↓, decreased. Joshua Stokes (St. Jude Children’s Research Hospital) assisted in the preparation of this figure.
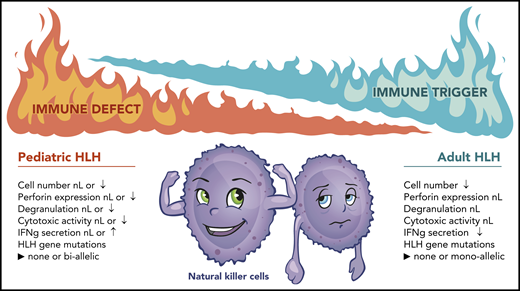
Immunologic and genetic differences between pediatric and adult HLH. nL, normal; ↑, increased; ↓, decreased. Joshua Stokes (St. Jude Children’s Research Hospital) assisted in the preparation of this figure.
HLH is a rare disorder of the immune system characterized by the excessive activation of lymphocytes and macrophages that copiously secrete proinflammatory cytokines. Although best known for its occurrence in young children, Scott and Robb-Smith first described HLH in a series of 4 adults who presented with fever, lymphadenopathy, hepatosplenomegaly, peripheral blood cytopenias, and pathologic evidence of cellular proliferation with accumulation of histiocytes, some of whom showed erythrophagocytosis.2 In 1952, a similar disorder affecting infant siblings was described, suggesting that, in some HLH cases, disease is caused by host genetic factors.3 Through these and other studies, it has since become clear that HLH occurs as a hereditary (also known as “primary”) disorder that is caused by pathogenic germline variants in critical immunoregulatory genes (many of which are involved in perforin-dependent cytotoxicity) or as a nonhereditary “secondary” disorder that is induced by strong immunologic triggers, including infections, malignancies, and autoimmune diseases. In immunocompetent individuals, cytotoxic lymphocytes, such as NK cells and CD8+ T cells, kill virus-infected and activated antigen-presenting cells and, thus, are critical for maintaining immune homeostasis.4 In contrast, in children with primary HLH, these processes are impaired, leading to a scenario in which even the slightest of triggers can bring on an exaggerated immune response marked by severe and often fatal tissue damage. Because of similarities in the clinical, laboratory, and histologic manifestations between pediatric and adult HLH, it has been questioned whether an analogous pathobiology occurs in both disorders.
To address this question, Carvelli et al systematically analyzed immune cell number and function in 68 adults with presumed secondary HLH and then correlated these findings with germline genetic information. To place the results from adult HLH patients into context, the investigators compared them with those obtained from a separate cohort of 34 individuals with various inflammatory diseases, as well as healthy volunteers. Importantly, none of the HLH patients or disease controls received immune suppression or other treatments prior to investigation. Therefore, the results obtained should reflect the native state of the immune cells being examined.
Through this study, the investigators make several interesting observations. In contrast with children with primary HLH, who often retain normal numbers of NK cells that display reduced cytotoxic function,5 most adult HLH patients exhibit a significant lymphopenia that resolves upon recovery from HLH. Despite their reduced numbers, NK cells from adults with HLH are indistinguishable from NK cells from disease or healthy control individuals in terms of perforin content and degranulation. Consistent with these findings, the investigators did not observe any defects in NK cell cytotoxicity after normalizing the percentage of NK cells among HLH, disease controls, and healthy individuals. The finding of normal cytotoxicity is perhaps not surprising when considered in light of the genetic results, which did not reveal any biallellic pathogenic variants in the 9 HLH-associated genes examined. Rather, 15 of 28 patients (50%) examined harbored ≥1 VUS, with most patients harboring only a single variant allele. Strikingly, the hypomorphic PRF1 A91V variant, which moderately affects protein stability and function, was present in half of the patients tested. These data are similar to a study by Zhang et al that identified germline HLH gene variants in 25 of 175 (14%) adult HLH patients examined, including a high prevalence of monoallelic variants, as well as the PRF1 A91V variant.6 Data from mouse models reveal that monoallelic variants in a single HLH gene do not reduce cytotoxicity or increase susceptibility to virus infection.7 Thus, it is unlikely that monoallelic VUS will impair NK cell cytotoxic activity in humans. In contrast, animals harboring increasing combinations of monoallelic Prf1, Stx11, and Lyst variants exhibit escalating impairments in cytotoxicity and virus-induced inflammation.7 Accordingly, the presence of multiple germline variants may confer a deleterious effect in response to a strong immunologic trigger.
Despite their normal cytotoxic activity, the number of IFN-γ–producing NK cells is significantly reduced in adults with HLH compared with healthy volunteers, but it is not different from that observed in adults with inflammatory diseases. Recently, Gao et al observed a similar defect in IFN-γ secretion by adult HLH NK cells and found that these cells expressed increased levels of the inhibitory receptor NKG2A and increased levels of PD-1, TIM-3, and LAG-3, suggesting that the NK cells were exhausted.8 It should be noted that the combination of increased NK cell activation, but reduced NK cell number and cytokine production, strongly resembles a phenotype previously reported in patients with sepsis.9,10 Altogether, these results intimate that, when placed in a hyperinflammatory setting, adult HLH NK cells become energized, yet exhausted (see figure). As a consequence, they can no longer properly respond to stimuli or control the immune response.
As with any good study, the report by Carvelli et al raises more questions than it answers. For example, does the reduced number of NK cells, despite their normal cytotoxic activity, contribute to the pathobiology of adult HLH, or is it simply a consequence? Is there a critical NK cell number below which HLH disease is more likely? What is the mechanism underlying the reduced NK cell IFN-γ secretion, and does it extend to other cytokines? Can this phenomenon be reversed to control inflammation? Toward this end, IFN-γ infusions are being studied as a possible therapy for sepsis-associated immunoparalysis (NCT01649921). Finally, should adults with HLH undergo routine immunologic or germline genetic testing? From the studies to date, it would seem prudent to examine perforin expression and degranulation in all adults with suspected HLH. If the results are abnormal, genetic testing is warranted to identify individuals who might have the primary form of disease. Although broader genetic testing in adults with HLH could be informative, this testing is best done in the context of a research protocol along with collection of detailed clinical and immunologic information. Only then will we better understand whether and how any identified abnormalities contribute to disease and influence outcomes.
Conflict-of-interest disclosure: K.E.N. and M.R.H. have received research funding from Incyte Corporation.

