How much clotting is enough to prevent bleeding is the ultimate question for treating bleeding disorders. Years of basic and clinical research have established the relationship between factor VIII (FVIII) replacement levels and bleeding risk, guiding the current practice in hemophilia. With the introduction of emicizumab, the bispecific ACE910 antibody, as a non-FVIII alternative to reduce bleeding in hemophilia A, the question of how much clotting is enough is at the forefront once again. In this issue of Blood, present an emicizumab-adapted hemophilia A mouse bleeding model that can help answer this question by enabling in vivo studies of emicizumab’s mechanisms of action and comparison with traditional FVIII replacement.1
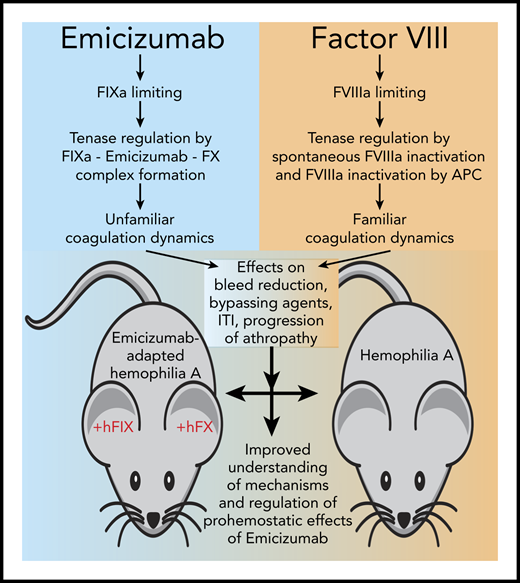
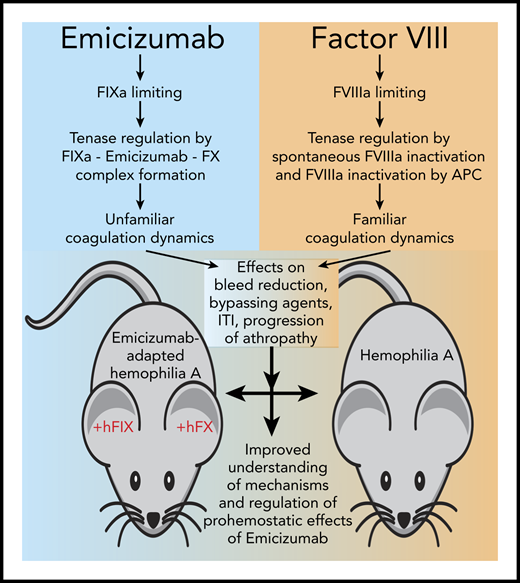
Research enabled by the emicizumab-adapted hemophilia A mouse bleeding model. Emicizumab (Hemlibra, also known as ACE910) is a FVIIIa mimetic that changes the familiar coagulation dynamics of FVIII replacement in hemophilia A since it is not based on FVIII. Instead, the activity of Emicizumab is regulated by the generation of FIXa and the formation of the heterodimeric FIX(a)-emicizumab-FX complex, while traditional regulation of FVIIIa by spontaneous inactivation and proteolytic inactivation by activated protein C (APC) are no longer applicable. This change in coagulation dynamics has several implications in hemophilia for the reduction of bleeding, the activity of other bypassing agents, and possibly the concentration of FVIII required for immune tolerance induction (ITI) and progression of hemophilic arthropathy (see text for details). The emicizumab-adapted hemophilia A mouse bleeding model, using human FIX (+hFIX) and FX (+hFX), enables comparison of the effects of emicizumab to that of FVIII to improve our understanding of the mechanisms and regulation of prohemostatic effects of emicizumab.
Research enabled by the emicizumab-adapted hemophilia A mouse bleeding model. Emicizumab (Hemlibra, also known as ACE910) is a FVIIIa mimetic that changes the familiar coagulation dynamics of FVIII replacement in hemophilia A since it is not based on FVIII. Instead, the activity of Emicizumab is regulated by the generation of FIXa and the formation of the heterodimeric FIX(a)-emicizumab-FX complex, while traditional regulation of FVIIIa by spontaneous inactivation and proteolytic inactivation by activated protein C (APC) are no longer applicable. This change in coagulation dynamics has several implications in hemophilia for the reduction of bleeding, the activity of other bypassing agents, and possibly the concentration of FVIII required for immune tolerance induction (ITI) and progression of hemophilic arthropathy (see text for details). The emicizumab-adapted hemophilia A mouse bleeding model, using human FIX (+hFIX) and FX (+hFX), enables comparison of the effects of emicizumab to that of FVIII to improve our understanding of the mechanisms and regulation of prohemostatic effects of emicizumab.
Individuals with severe hemophilia A, the genetic deficiency of coagulation FVIII (FVIII ≤1%), are at the greatest risk for severe bleeding, which often involves repeated bleeding episodes in weight-bearing joints that when left untreated progress into hemophilic arthropathy, a debilitating joint disease that greatly affects quality of life.2 The current clinical paradigm to maintain FVIII levels >1% greatly reduces bleeding risk, particularly in the joints, but requires frequent and lifelong administration of FVIII. Furthermore, the development of inhibitory antibodies to FVIII in ∼25% to 30% of individuals on replacement therapy renders them at high risk for bleeding requiring alternative (bypassing) agents to prevent and treat bleeding.3 These limitations of FVIII have spurred the development of improved FVIII molecules and new bypassing strategies that include emicizumab.
Emicizumab (Hemlibra; also known as ACE910) is a heterodimeric antibody interacting with both FIX(a) and FX, thereby mimicking a major function of FVIIIa to bring FIXa in close proximity to FX to promote its activation to FXa.4 Several clinical trials have demonstrated potent prohemostatic effects of emicizumab in hemophilia A with and without inhibitors.5-7 Since emicizumab may be administered as infrequently as once every 4 weeks, it addresses 2 important limitations of FVIII replacement therapy, namely inhibitor formation and the emotional and physical stress associated with multiple weekly injections. While this is great news for individuals with hemophilia A as the therapeutic repertoire expands, it also presents a number of difficult questions related to the extent and situations in which emicizumab may replace traditional FVIII replacement therapy. Typically, answers to such questions are found and supported by extensive experimental data, but experimental in vivo support has been limited to nonhuman primate models due to the specificity of emicizumab for human (and primate) FIX(a) and FX.8 For instance, the seemingly straightforward question “What is the FVIII equivalence of emicizumab?” has been proven difficult to answer.9
Ferrière et al created a workaround for the specificity limitation of emicizumab by injecting human FIX and FX in the hemophilia A mouse shortly before the induction of bleeding. Using this emicizumab-adapted hemophilia A mouse bleeding model, the FVIII equivalence of emicizumab is found to be 9 U/dL in a physiological environment,1 thereby supporting the earlier notion that emicizumab changes the phenotype of severe hemophilia A to resemble a moderate (1% to 5% FVIII) to mild (5% to 40% FVIII) phenotype.9
This begs the question of how much clotting is enough. It is important to note that in the presence of Emicizumab traditional regulation of the tenase complex by spontaneous dissociation of FVIIIa or inactivation of FVIIIa by activated protein C are no longer applicable (see figure). Instead, the procoagulant activity of emicizumab is regulated by the availability of FIXa and the equilibrium constants for the formation of the emicizumab-FIX(a)-FX complex.4,9 As a result, the clotting dynamics of emicizumab are different from what we are used to, which is perhaps best illustrated by the on/off bleed reduction without a typical dose-response effect of emicizumab in the mouse model.1 This illustrates the need for a more in-depth understanding how the activity of emicizumab is regulated and its mechanism of action. The availability of an emicizumab-adapted mouse model contributes importantly to obtain such understanding.
In addition to the afore mentioned question how emicizumab and FVIII compare, another important question is how to treat breakthrough bleeds in the presence of emicizumab. From the clinical trial experience,6 it is clear that adjustments of standard bypassing therapy are needed, and the mouse model can help navigate these new clotting dynamics to find the balance between efficacy and safety. Another area where the mouse model can provide unique insights is for immune tolerance induction to eradicate inhibitors and how emicizumab may help to reduce the required FVIII dose, which is associated with enormous cost.10 The observation that FVIII provides additional prohemostatic effects in the presence of emicizumab in the mouse model is therefore noteworthy.1 Key for any prohemostatic therapy in hemophilia is how the development of hemophilic arthropathy is affected. Early experimental data in monkeys indicate that emicizumab prevents joint bleeds,8 but clinical trial data indicate that emicizumab does not reduce joint bleeds to 0 in all patients (and neither does FVIII)2,5-7 ; thus, a better understanding of the effect of emicizumab on the progression and management of hemophilic arthropathy is urgently needed. The emicizumab-adapted hemophilia A mouse model will encompass an important tool to obtain such insights. However, it should be noted that additional modifications are needed, as indicated, before this model is suited for longer-term hemophilic arthropathy studies.1
Finally, while it is typical to address bleeding in hemophilia from a clotting-centric perspective, it is equally important not to overlook that bleeding, and especially joint bleeding, has its own contributing mechanisms that in addition to coagulation may include endogenous anticoagulant pathways, fibrinolysis, vascular and bone remodeling pathways, and others. Joint bleeding is the cumulative disbalance of these pathways, and the mouse is arguable the best model to test how the contributions of these pathways are affected by emicizumab. The emicizumab-adapted hemophilia A mouse bleeding model developed by Ferrière et al enables such studies and is likely to stimulate new areas of hemophilia A research focused on emicizumab.
Conflict-of-interest disclosure: The Scripps Research Institute holds intellectual property rights with L.O.M. listed as inventor. L.O.M. is a cofounder and a member of the board of directors of Hematherix, a biotech company that is developing super-FVa therapy for bleeding complications.