In this issue of Blood, show that signal recognition particle 54 (SRP54) mutations found in patients with congenital neutropenia act in a dominant-negative manner on the functionality of the wild-type SRP54, leading to neutropenia because of impaired splicing of the transcription factor X-box binding protein 1 (XBP1).1
Congenital neutropenias are a heterogenous group of rare hematological disorders characterized mainly by impaired maturation of neutrophils.2 They are bone marrow failure syndromes presenting with severe neutropenia at birth owing to a myeloid maturation block at the promyelocyte/myelocyte stage. Patients with congenital neutropenia have recurrent infections from their first months of life, but the symptoms may ameliorate later in life. In severe cases, symptoms in other organs, such as the pancreas, central nervous system, heart, bone, and skin, may occur. Rigorous studies using exome sequencing have identified mutations in ∼25 different genes.3 Mutations in ELANE genes account for ∼50% of the cases4 ; however, mutations are also present in other genes, like HAX1, G6PC3, SRP54, GFI, and CSF3R. Although many mutated genes implicated in the disease have been identified, we still lack knowledge of the function and the downstream mechanisms that are affected by these mutations. One solution to this problem is to create animal models that will permit assessment of the phenotypic abnormalities caused by specific mutations and that will offer the tools to study the molecular mechanism responsible for the phenotypes. For instance, mouse models represent a wonderful tool for dissecting the function of specific mutations.5 However, both the cost and the number of animals needed preclude wide use of a murine model. Zebrafish, on the other hand, are easy to genetically manipulate, and the relatively low cost makes it ideal for testing multiple mutations and even to establish a “personalized approach.” Successful models for congenital neutropenia have been established in zebrafish, showing, for example, that Hax1 deficiency impairs neutrophilic development.6 Schürch et al have now characterized a zebrafish line that lacks Srp54 and exhibits low numbers of neutrophils. However, systemic effects lead to its lethality at embryonic stages. Surprisingly, the complementation of the heterozygous-viable animals with mutated SRP54 revealed the dominant-negative role of these mutations.
Mutations on the SRP54 gene have been identified relatively recently in patients with congenital neutropenia and have been sometimes associated with neurodevelopmental delay and/or an exocrine pancreatic insufficiency.7,8 Patients with such mutations exhibit reduced proliferation of granulocytes accompanied by endoplasmic reticulum (ER) stress.7 Zebrafish deficient for srp54 by means of morpholino injection exhibited significant reduction in neutrophil counts and impaired chemotaxis, but also decreased size of the exocrine pancreas.8 The mutant zebrafish studied here by Schürch et al recapitulated the majority of these phenotypes. Indeed, srp54sa11820/sa11820 mutants display impaired development concomitant with multiple phenotypic abnormalities in many organs, leading to their embryonic lethality. Heterozygous and homozygous animals present lower numbers of neutrophils, as measured by in situ hybridization and cell sorting experiments. However, the phenotype is much milder in the heterozygous animals. Interestingly, the symptoms are alleviated in 2-year-old heterozygous fish, recapitulating the human phenotype. The size of the exocrine pancreas was similar between control and heterozygous animals, but the early lethality of the homozygous animals precluded analysis of the pancreas. Importantly, injection of human SRP54 messenger RNA (mRNA) could transiently rescue the neutropenia and the lethal phenotype of null embryos. Further investigation of the role of actual patient mutations led to intriguing results. Although injection of mutant mRNA could not rescue or only partially rescue embryonic lethality in srp54sa11820/sa11820 mutants, injection to heterozygous animals aggravated their phenotype and led to a severely reduced number of neutrophils and reduced size of the exocrine pancreas. These results suggested a dominant-negative function of the mutations, which was verified in primary human cells and cell lines. These experiments not only showed the dominant-negative function of the mutants, which could be easily missed in studies of knockout animals, but also clarified that the severity of the disease varies depending on the mutations. Thus, diverse dominant-negative effects exercised by different mutations readily explain the phenotypic variability observed in patients.
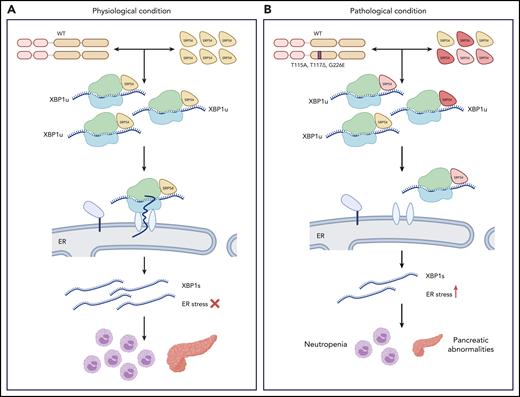
Next, the authors investigated the mechanistic underpinnings of the observed phenotypes. SRP54 is part of the signal recognition particle ribonucleoprotein complex responsible for the cotranslational targeting of proteins to the ER under conditions of ER stress. This targeting is crucial for activation of the unfolded protein response that is needed to restore ER homeostasis.9 XBP1 is a mediator of the unfolded protein response activated by splicing at the ER membrane. Its translocation to the ER membrane is dependent on SRP54. Notably, granulopoiesis has been highly linked to splicing dysfunction.10 Thus, the authors hypothesized that defective xbp1 splicing may lie downstream of the SRP54 mutations. Indeed, Xbp1-deficient embryos exhibited reduction in neutrophils and exocrine pancreas size, thus simulating the phenotypes of Srp54-deficient embryos. Induction of ER stress in srp54sa11820/sa11820 or srp54+/sa11820 mutants injected with mutated mRNA led to defective xbp1 splicing compared with wild-type or srp54+/sa11820 animals and reduced the number of neutrophils compared with control treatment. Importantly, these phenotypes were conserved in human cells. Ultimately, spliced, but not unspliced xbp1, could rescue the neutrophil numbers of srp54sa11820/sa11820 embryos, verifying that impaired xbp1 splicing is one of the causes of the neutropenic phenotype downstream of SRP54 mutations (see figure).

Impaired XBP1 splicing downstream of SRP54 mutations. During physiological conditions (A), the signal recognition particle 54 (SRP54) facilitates the translocation of the ribosome-associated unspliced XBP1 (XBP1u) to the ER where it is spliced (XBP1s). In pathological conditions (B), SRP54 mutations found in patients act in a dominant-negative manner to wild-type (WT) SRP54 and impair XBP1 splicing, leading to neutropenia and decreased size of the exocrine pancreas. Image by Veronica Bergo, created with BioRender.com.
Impaired XBP1 splicing downstream of SRP54 mutations. During physiological conditions (A), the signal recognition particle 54 (SRP54) facilitates the translocation of the ribosome-associated unspliced XBP1 (XBP1u) to the ER where it is spliced (XBP1s). In pathological conditions (B), SRP54 mutations found in patients act in a dominant-negative manner to wild-type (WT) SRP54 and impair XBP1 splicing, leading to neutropenia and decreased size of the exocrine pancreas. Image by Veronica Bergo, created with BioRender.com.
Collectively, this important and timely study revealed the dominant-negative function of SPR54 mutations. This information may be used to refine and better direct medical treatments. The paper also emphasizes the value of animal models and the need to examine the mechanistic implications of every single mutation to achieve a “personalized approach” for patients with congenital neutropenia.
Conflict-of-interest disclosure: The author declares no competing financial interests.