Key Points
Dual inhibition of MCL1 and BCL2 proteins results in a durable remission in DLBCL mouse models.
S63845 or venetoclax encapsulated in tumor-targeted nanoparticles improves the therapeutic index of the combination.

Abstract
Cancer and normal cells use multiple antiapoptotic BCL2 proteins to prevent cell death. Therapeutic targeting of multiple BCL2 family proteins enhances tumor killing but is also associated with increased systemic toxicity. Here, we demonstrate that the dual targeting of MCL1 and BCL2 proteins using the small molecules S63845 and venetoclax induces durable remissions in mice that harbor human diffuse large B-cell lymphoma (DLBCL) tumors but is accompanied by hematologic toxicity and weight loss. To mitigate these toxicities, we encapsulated S63845 or venetoclax into nanoparticles that target P-selectin, which is enriched in tumor endothelial cells. In vivo and ex vivo imaging demonstrated preferential targeting of the nanoparticles to lymphoma tumors over vital organs. Mass spectrometry analyses after administration of nanoparticle drugs confirmed tumor enrichment of the drug while reducing plasma levels. Furthermore, nanoparticle encapsulation allowed 3.5- to 6.5-fold reduction in drug dose, induced sustained remissions, and minimized toxicity. Our results support the development of nanoparticles to deliver BH3 mimetic combinations in lymphoma and in general for toxic drugs in cancer therapy.
Introduction
Dysregulated expression or function of members of the BCL2 protein family has been implicated in carcinogenesis, including lymphomagenesis, and resistance to a variety of anticancer drugs.1-3 Modulating the expression level or function of some of these proteins is currently under intensive investigation for cancer therapy.4,5 Because both cancer and normal cells use multiple antiapoptotic proteins to promote their survival, it is not surprising that earlier attempts to target more than 1 protein, such as BCL2 and BCL-xL, were associated with increased toxicity.6
We and others have recently demonstrated the codependency of lymphoma cells on BCL2 and MCL1, suggesting that dual inhibition of both proteins will be necessary to increase the therapeutic outcome.7-9 Synergistic interaction between the MCL1 selective inhibitor S63845 and the BCL2 selective inhibitor venetoclax has been demonstrated in preclinical models of solid tumors and hematologic malignancies.10-18 However, the potential toxicity of dual inhibition of MCL1 and BCL2, which have critical prosurvival roles in several organs, may limit their systemic administration to patients with cancer. Venetoclax is approved for use in patients with chronic lymphocytic leukemia (CLL) and small lymphocytic leukemia (SLL),19 and its safety profile is known.20 It is mostly well tolerated, but a recent analysis of 350 CLL patients receiving venetoclax monotherapy revealed the following hematologic adverse events: 37% had neutropenia, 31% had anemia and 14% had thromobocytopenia.20 This is not surprising, given the well-established role of BCL2 in several stages of hematopoietic development as well as adult hematopoietic maintenance.21 The safety and potential efficacy of MCL1 inhibitors are currently unknown. Ubiquitous knockout of MCL1 leads to peri-implantation embryonic lethality in mice.22 Conditional knockout studies in mice have demonstrated the critical dependence of hematopoietic stem cells and B and T lymphocytes in various stages of development on MCL1.23-25 More recently, MCL1 inhibition has been shown to induce cardiomyocyte toxicity.26 It has also been demonstrated that in the hematopoietic system, BCL2 proteins may act alone or redundantly20 ; further caution regarding toxicity should be used when multiple members of the of BCL2 protein family are inhibited simultaneously.
Improving the therapeutic index of combinations of BCL2 protein family inhibitors will be required for the successful development of this strategy for cancer therapy. The essentiality of MCL1 in normal cells can underlie the significant toxicities of such combinations. To combat the toxicity of small molecule drugs in normal organs, targeted nanoparticle delivery methods have been used to effectively maximize drug concentrations at tumor sites while reducing the exposure of vital organs to drugs.27 Nanoparticles are submicron-size particles up to several hundred nanometers (nm) in size that can be used to encapsulate drugs to prevent the unbiased penetration of the drug upon systemic administration. Nanoparticles can be customized for the delivery goal and coated with a tumor-targeting agent such as a protein or polysaccharide.27-30 Despite the potential for nanodelivery systems to dramatically improve the therapeutic indices of their cargoes, this approach is often overlooked until severe toxicities are observed in the clinic. Through a multidisciplinary design that connects cancer biology and pharmacology to nanotechnology, it is possible to anticipate potential issues and solve them in preclinical studies, thus enabling safer, more efficient translation to patients.
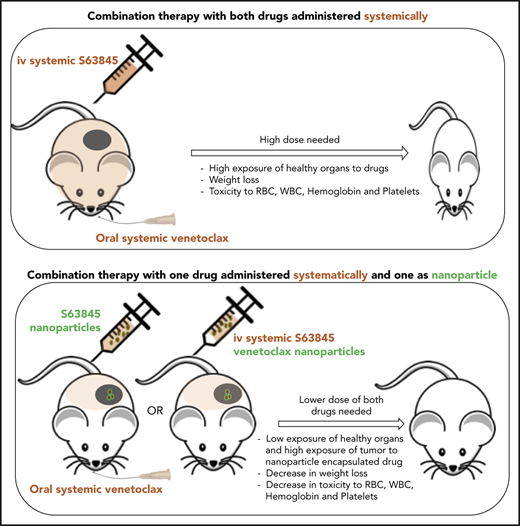
In this article, we present a highly efficacious targeted delivery strategy for the dual inhibition of MCL1 and BCL2 in lymphoma. Our nanoparticle delivery approach involves a simple vehicle that enables high drug loading and encapsulation efficiency of the cargo. First, we report that the dual inhibition of MCL1 (by S63845) and BCL2 (by venetoclax) by using traditional systemic intravenous and/or oral approaches resulted in a durable remission in diffuse large B-cell lymphoma (DLBCL) mouse models but was also associated with significant hematologic toxicity and weight loss. To resolve this issue, we present new formulation strategies for tumor-targeting nanoparticles that encapsulate either S63845 or venetoclax. Administration of 1 nanoformulation in combination with the other free drug (S63845 nanoparticles plus free venetoclax or free S63845 plus venetoclax nanoparticles) improved the therapeutic index of the combination. Specifically, this approach enabled a 50% decrease in the drug dose and reduced toxicity while maintaining tumor-killing efficacy in vivo. To our knowledge, this is the first study to demonstrate the advantage of targeted nanoparticle delivery methods of BH3 mimetics in lymphoma.
Methods
Cell lines and reagents
Human DLBCL-derived cell lines SU-DHL-6, OCI-Ly19, U-2973, SU-DHL-4, SU-DHL-8, Ri-1, and U-2932, and the MCL-derived cell line Maver-1 were obtained from the DSMZ-German Collection of Microorganisms and Cell Cultures, Department of Human and Animal Cell Cultures (Braunschweig, Germany). Cell lines TMD8 and HBL-1 were kindly provided by R. E. Davis (MD Anderson Cancer Center, Houston, TX). Human MCL-derived cell lines Mino, Jeko-1, Rec-1, and Z-138 were obtained from American Type Culture Collection. All cell lines were authenticated using a targeted deep sequencing assay of 585 cancer genes (HemePACT) at the Integrated Genomic Operation Core (Memorial Sloan Kettering Cancer Center). Mutational landscapes for the cell lines were characterized and are available on the cBioportal platform at https://www.cbioportal.org/study/summary?id=lymphoma_cellline_msk_2020. Cell lines were cultured in RPMI 1640 medium supplemented with 10% to 20% heat-inactivated fetal bovine serum (Hyclone, SH30396.03, GE Healthcare Life Sciences) and 1% l-glutamine and penicillin-streptomycin in a humid environment of 5% CO2 at 37°C. Cell lines were tested for mycoplasma before being used in any mouse experiment. Routine mycoplasma testing for all cell lines was performed every 6 months. S63845 (C-1370) was purchased from Chemgood (Glen Allen, VA). A1210477 (A-9036) was purchased from Active Biochem (Kowloon, Hong Kong). UMI-77 (S7531) and venetoclax (S8048) were purchased from Selleckchem (Houston, TX).
Nanoparticle preparation and characterization
Nanoparticle formulation conditions were determined with a predictive model previously published.31 Briefly, logP, the number of high intrinsic state substructures, and the pKa of the most acidic and basic groups of each drug were determined and used to determine the dye partner and pH of the nanoprecipitation and resuspension conditions. Either S63845 or venetoclax was dissolved in dimethyl sulfoxide at a concentration of 20 mg/mL. An aqueous solution consisting of 225 μL of double-distilled water (ddH2O), 225 μL of fucoidan (20 mg/mL in ddH2O), and 100 μL of IR-783 (2 mg/mL in ddH2O) was prepared. Fifty microliters of drug was added dropwise to this aqueous solution under agitation on a vortex mixer. This step was followed by a 10-minute centrifugation at 10 000 relative centrifugal force [RCF] at room temperature. The nanoparticle pellet was resuspended in 200 μL of ddH2O for S63845 or sterile 1× phosphate-buffered saline (PBS) for venetoclax. Encapsulation efficiency was quantified using high-performance liquid chromatography. Dynamic light scattering and ζ potential measurements were conducted in PBS using a Zetasizer Nano ZS size analyzer (Malvern). Dynamic light scattering was conducted with nanoparticles in PBS, and ζ potential was measured in sterile water to prevent interactions of buffer ions with the nanoparticle surface. The mean loading efficiency for venetoclax nanoparticles is 43.84% and 19.02% for S63845 nanoparticles.
Statistics
Results are expressed as the mean from at least 3 independent experiments or biological replicates as indicated in the Figure legends. Statistical significance for difference in protein expression levels in resistant and sensitive cell lines was measured by an unpaired Student t test. Tumor volume is represented as means, and error bars represent standard deviation (SD). Two-way analysis of variance was used to determine statistical significance between the first day of treatment and subsequent time points for tumor regression as well as weight loss analyses. Statistical significance for difference in ex vivo fluorescence relative to vehicle was calculated by Mann-Whitney U test. For hematologic toxicity, 1-way analysis of variance was used to determine statistical significance between the first day of treatment and subsequent time points. All statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software). A value of P < .05 was considered statistically significant.
Results
Activity of the selective MCL1 inhibitor S63845 is enhanced in combination with the BCL2 inhibitor venetoclax
We examined the single-agent activity of S63845 in a panel of 14 lymphoma cell lines representing different cells of origin and differentiation states (Figure 1A). In 4 cell lines, the half maximal inhibitory concentration (IC50) was <0.1 µM (termed sensitive cell lines), 5 cell lines were moderately sensitive (IC50 between 0.1 and 1 µM), and 5 cell lines were resistant (IC50 >1 µM). Only 4 cell lines (29%) were sensitive to both MCL1 and BCL2 inhibitors (Figure 1A). Cell lines that were sensitive to S63845 expressed lower levels of the antiapoptotic proteins MCL1, BCL2, and BCL-xL and the pro-apoptotic protein BIM (Figure 1B; supplemental Figure 1A [available on the Blood Web site]). Sensitivity to S63845 was observed irrespective of a variety of genetic alterations in BCL2 family members, including TP53 (supplemental Figure 1B) (genetic alterations are available at https://www.cbioportal.org/study/summary?id=lymphoma_cellline_msk_2020).
![S63845 has antiproliferative activity in lymphoma cells and synergizes with venetoclax. (A) Heat map summarizing the IC50 (mean ± standard error of the mean [SEM]) in µM of S63845 and venetoclax in 14 lymphoma cell lines with the following subtypes: activated B-cell (ABC) DLBCL, germinal center B-cell (GCB) DLBCL, double-hit lymphoma (DHL), and mantle cell lymphoma (MCL). Cells were incubated with increasing concentrations of drug for 72 hours. Data represent the mean of 3 separate experiments. Cell viability was assessed by CellTiter-Glo. Sensitive, IC50 <0.1 µM; moderately sensitive, IC50 between 0.1 and 1 µM; resistant, IC50 >1 µM. (B) Dot plots quantifying MCL1, BCL2, BCL-xL, and BIM protein expression from western blot in supplemental Figure 2A in the 4 cell lines most resistant (Res) (U-2932, HBL-1, Ri-1, and Maver-1) and sensitive (Sen) (U-2973, OCI-LY19, Mino, and SU-DHL-6) to S63845. Each point represents the protein:β-actin ratio in a cell line. Error bars represent SD from 4 different cell lines listed above. Statistical significance was measured by an unpaired Student t test. (C) Heat map summarizing the effect of combinations of varying concentrations of S63845 and venetoclax on cell viability in the indicated cell lines as assessed by the CellTiter-Glo assay. Combination responses are examined in a 5 × 5 viability matrix after 24 hours of treatment. Percentage of cell viability is depicted in a colorimetric scale from black (high) to white (low) normalized to dimethyl sulfoxide control. Values are the mean of 3 separate experiments. (D) Box plot graph summarizing the results of treatment with combined S63845 and venetoclax for 24 hours in the 3 cell lines most resistant to S63845 (Ri-1, HBL-1, and U-2932) and venetoclax (TMD8, Z-138, and Jeko-1). The y-axis depicts the log-odds that indicate the ratio between observed and expected inhibition on a log scale. Log-odds >0 signify antagonism, log-odds of 0 signify an additive effect, and log-odds <0 (area below gray line) indicate synergy between the 2 drugs. Cell viability was assessed by CellTiter-Glo. All data points represent the mean of 3 separate experiments. ***P < .001; **P < .01; *P = .02.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/15/10.1182_blood.2020008017/2/m_bloodbld2020008017f1.png?Expires=1769402102&Signature=bvOk40aPh7r980h4144IAB3F0U6b8dR1YXmB3cFLZw5FtpXplGVbkNdoTleBTMVdSAmndzvVZ345DPCOaPayun7Xr1CWKgOuAvvj69E6zXKhYlDD50qZce9NR~I9-djINNoo6k9TUxWlG2iloeEZe8kswER~UgptHn-LlcuaBqRY6h5QFgzvFiUKkPQyJeygzuQMh51PI3j~1~EpbtwWe~RAxM6j1gd2y4hJNsI7qYKl-6Prz5BEEeEEZOYLnaYpR5IEqiu6a-emPIJneEnItXRnZuhkyH40ju08fcrYj5AQ~TebV6YqA3x1UlwUkqyTZu78U5dpDcuHGvSW-41e7g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
S63845 has antiproliferative activity in lymphoma cells and synergizes with venetoclax. (A) Heat map summarizing the IC50 (mean ± standard error of the mean [SEM]) in µM of S63845 and venetoclax in 14 lymphoma cell lines with the following subtypes: activated B-cell (ABC) DLBCL, germinal center B-cell (GCB) DLBCL, double-hit lymphoma (DHL), and mantle cell lymphoma (MCL). Cells were incubated with increasing concentrations of drug for 72 hours. Data represent the mean of 3 separate experiments. Cell viability was assessed by CellTiter-Glo. Sensitive, IC50 <0.1 µM; moderately sensitive, IC50 between 0.1 and 1 µM; resistant, IC50 >1 µM. (B) Dot plots quantifying MCL1, BCL2, BCL-xL, and BIM protein expression from western blot in supplemental Figure 2A in the 4 cell lines most resistant (Res) (U-2932, HBL-1, Ri-1, and Maver-1) and sensitive (Sen) (U-2973, OCI-LY19, Mino, and SU-DHL-6) to S63845. Each point represents the protein:β-actin ratio in a cell line. Error bars represent SD from 4 different cell lines listed above. Statistical significance was measured by an unpaired Student t test. (C) Heat map summarizing the effect of combinations of varying concentrations of S63845 and venetoclax on cell viability in the indicated cell lines as assessed by the CellTiter-Glo assay. Combination responses are examined in a 5 × 5 viability matrix after 24 hours of treatment. Percentage of cell viability is depicted in a colorimetric scale from black (high) to white (low) normalized to dimethyl sulfoxide control. Values are the mean of 3 separate experiments. (D) Box plot graph summarizing the results of treatment with combined S63845 and venetoclax for 24 hours in the 3 cell lines most resistant to S63845 (Ri-1, HBL-1, and U-2932) and venetoclax (TMD8, Z-138, and Jeko-1). The y-axis depicts the log-odds that indicate the ratio between observed and expected inhibition on a log scale. Log-odds >0 signify antagonism, log-odds of 0 signify an additive effect, and log-odds <0 (area below gray line) indicate synergy between the 2 drugs. Cell viability was assessed by CellTiter-Glo. All data points represent the mean of 3 separate experiments. ***P < .001; **P < .01; *P = .02.
S63845 has antiproliferative activity in lymphoma cells and synergizes with venetoclax. (A) Heat map summarizing the IC50 (mean ± standard error of the mean [SEM]) in µM of S63845 and venetoclax in 14 lymphoma cell lines with the following subtypes: activated B-cell (ABC) DLBCL, germinal center B-cell (GCB) DLBCL, double-hit lymphoma (DHL), and mantle cell lymphoma (MCL). Cells were incubated with increasing concentrations of drug for 72 hours. Data represent the mean of 3 separate experiments. Cell viability was assessed by CellTiter-Glo. Sensitive, IC50 <0.1 µM; moderately sensitive, IC50 between 0.1 and 1 µM; resistant, IC50 >1 µM. (B) Dot plots quantifying MCL1, BCL2, BCL-xL, and BIM protein expression from western blot in supplemental Figure 2A in the 4 cell lines most resistant (Res) (U-2932, HBL-1, Ri-1, and Maver-1) and sensitive (Sen) (U-2973, OCI-LY19, Mino, and SU-DHL-6) to S63845. Each point represents the protein:β-actin ratio in a cell line. Error bars represent SD from 4 different cell lines listed above. Statistical significance was measured by an unpaired Student t test. (C) Heat map summarizing the effect of combinations of varying concentrations of S63845 and venetoclax on cell viability in the indicated cell lines as assessed by the CellTiter-Glo assay. Combination responses are examined in a 5 × 5 viability matrix after 24 hours of treatment. Percentage of cell viability is depicted in a colorimetric scale from black (high) to white (low) normalized to dimethyl sulfoxide control. Values are the mean of 3 separate experiments. (D) Box plot graph summarizing the results of treatment with combined S63845 and venetoclax for 24 hours in the 3 cell lines most resistant to S63845 (Ri-1, HBL-1, and U-2932) and venetoclax (TMD8, Z-138, and Jeko-1). The y-axis depicts the log-odds that indicate the ratio between observed and expected inhibition on a log scale. Log-odds >0 signify antagonism, log-odds of 0 signify an additive effect, and log-odds <0 (area below gray line) indicate synergy between the 2 drugs. Cell viability was assessed by CellTiter-Glo. All data points represent the mean of 3 separate experiments. ***P < .001; **P < .01; *P = .02.
Venetoclax is a potent and selective BCL2 inhibitor approved for use in patients with CLL and SLL.19 Because 57% of the cell lines differentially responded to either S63845 or venetoclax (Figure 1A), we examined whether dual inhibition of BCL2 and MCL1 is more effective. In the cell lines assessed, venetoclax enhanced the efficacy of S63845, including in a cell line very resistant to both drugs (Figure 1C; supplemental Figure 2A). The combination even demonstrated enhanced activity in the 3 cell lines most resistant to S63845 (U-2932, HBL-1, and Ri-1) or venetoclax (Jeko-1, TMD8, and Z-138) (Figure 1D).
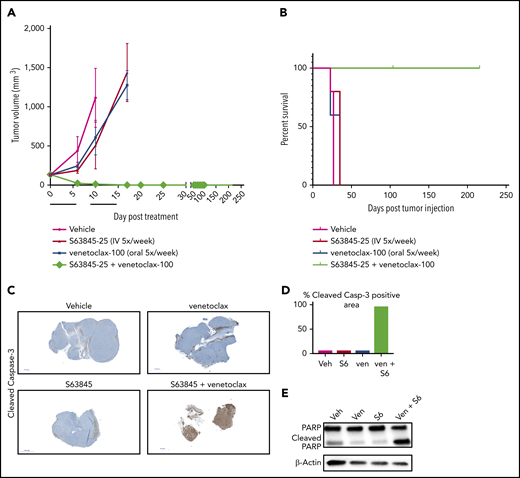
To evaluate the efficacy of the combination of S63845 and venetoclax in vivo, we generated a human DLBCL xenograft model using the SU-DHL-6 cell line (Figure 2A). Monotherapy with S63845 at 25 mg/kg administered intravenously (IV) or venetoclax administered at 100 mg/kg orally 5 times per week had a modest effect on tumor volume (tumor volume decrease of 41% upon S63845 monotherapy and 36% upon venetoclax monotherapy). However, a major impact was observed in mice administered a combination of S63845 and venetoclax: after only 9 treatments, we observed complete remission of tumors, which was maintained for up to 216 days (Figure 2A) and increased survival (Figure 2B). Immunohistochemistry and western blot analyses of tumors from different treatment groups revealed that treatment with each drug alone resulted in minimal cleavage of caspase 3 and its target poly(ADP-ribose) polymerase (PARP). In contrast, the combination of S63845 and venetoclax resulted in cleavage of both diffuse caspase 3 (Figure 2C-D) and PARP (Figure 2E), indicative of apoptosis.
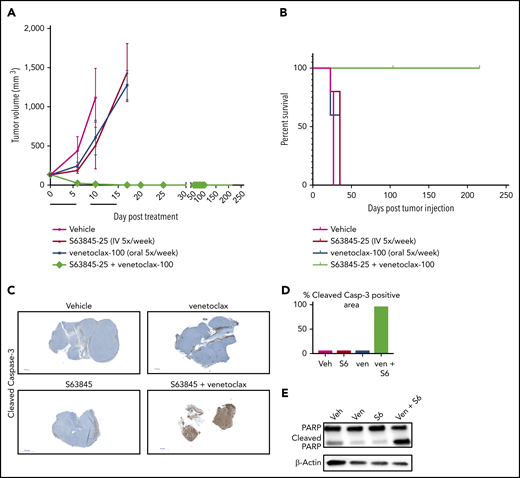
S63845 and venetoclax synergize in vivo and activate apoptosis. (A) DLBCL cell line SU-DHL-6 was xenografted subcutaneously into NSG female mice. After 10 days, mice were randomly assigned into comparable groups (5 mice each) and treated with vehicle (Veh), venetoclax (Ven) (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), and a combination of venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week) for 2 weeks (indicated by black line). Tumor volumes are represented as means; error bars represent SD. (B) Kaplan-Meier survival curve (survival percentage) of tumor-bearing mice treated as specified in panel A. (C) Immunohistochemistry (IHC) images at original magnification ×40 of mouse tumors from SU-DHL-6 xenografts exposed to different treatment groups described in panel A stained for cleaved caspase-3. (D) Quantification of cleaved caspase-3–positive cells in IHC images of mouse tumors in panel C. (E) Western blot analysis of protein derived from tumors to assess the effects of different drug treatments indicated in panel A. In vivo protein levels of poly (ADP-ribose) polymerase (PARP) and cleaved PARP were assessed on day 5 of treatment. β-Actin was used as a loading control.
S63845 and venetoclax synergize in vivo and activate apoptosis. (A) DLBCL cell line SU-DHL-6 was xenografted subcutaneously into NSG female mice. After 10 days, mice were randomly assigned into comparable groups (5 mice each) and treated with vehicle (Veh), venetoclax (Ven) (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), and a combination of venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week) for 2 weeks (indicated by black line). Tumor volumes are represented as means; error bars represent SD. (B) Kaplan-Meier survival curve (survival percentage) of tumor-bearing mice treated as specified in panel A. (C) Immunohistochemistry (IHC) images at original magnification ×40 of mouse tumors from SU-DHL-6 xenografts exposed to different treatment groups described in panel A stained for cleaved caspase-3. (D) Quantification of cleaved caspase-3–positive cells in IHC images of mouse tumors in panel C. (E) Western blot analysis of protein derived from tumors to assess the effects of different drug treatments indicated in panel A. In vivo protein levels of poly (ADP-ribose) polymerase (PARP) and cleaved PARP were assessed on day 5 of treatment. β-Actin was used as a loading control.
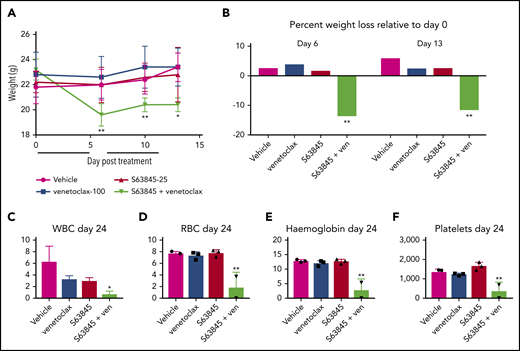
Mice treated with the S63845-venetoclax combination suffered significant weight loss of up to ∼15% of their original body weight; this was not observed in mice treated with each drug alone (Figure 3A-B). Similarly, the combination regimen was more toxic to peripheral blood white blood cells (WBCs) and red blood cells (RBCs) than either drug alone (Figure 3C-F). Collectively, these data suggest that the doses of S63845 (25 mg/kg 5 times per week) and venetoclax (100 mg/kg 5 times per week) needed for a sustained and durable remission are accompanied by significant toxicity.
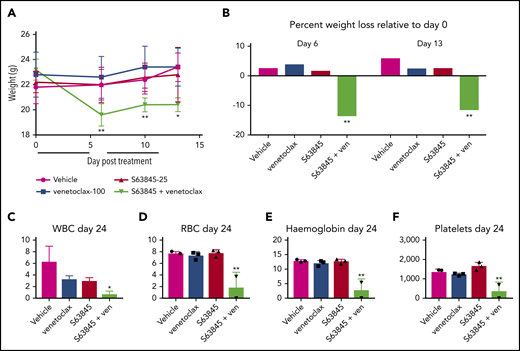
The dose of S63845 plus venetoclax needed for sustained remission is toxic. (A) Graph depicting weight variation posttreatment in SU-DHL-6 xenografts of different treatment groups: vehicle, venetoclax (ven) (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), and a combination of venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week) for 2 weeks (indicated by black line) (n = 5). Weights are represented as means; error bars represent SD. Two-way analysis of variance (ANOVA) was used to determine statistical significance between the first day of treatment and subsequent time points. (B) Histogram depicting weight variation on days 6 and 13 relative to day 0 posttreatment in different treatment groups from panel A. Two-way ANOVA was used to determine statistical significance between the first day of treatment and subsequent time points. (C-F) Analysis of the effect of combination therapy on WBCs (C), RBCs (D), hemoglobin (E), and platelets (F) on day 24 of treatment in mice from treatment groups detailed in panel A (n = 3). Error bars represent SD. One-way ANOVA was used to determine statistical significance relative to vehicle. **P < .01; *P < .05.
The dose of S63845 plus venetoclax needed for sustained remission is toxic. (A) Graph depicting weight variation posttreatment in SU-DHL-6 xenografts of different treatment groups: vehicle, venetoclax (ven) (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), and a combination of venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week) for 2 weeks (indicated by black line) (n = 5). Weights are represented as means; error bars represent SD. Two-way analysis of variance (ANOVA) was used to determine statistical significance between the first day of treatment and subsequent time points. (B) Histogram depicting weight variation on days 6 and 13 relative to day 0 posttreatment in different treatment groups from panel A. Two-way ANOVA was used to determine statistical significance between the first day of treatment and subsequent time points. (C-F) Analysis of the effect of combination therapy on WBCs (C), RBCs (D), hemoglobin (E), and platelets (F) on day 24 of treatment in mice from treatment groups detailed in panel A (n = 3). Error bars represent SD. One-way ANOVA was used to determine statistical significance relative to vehicle. **P < .01; *P < .05.
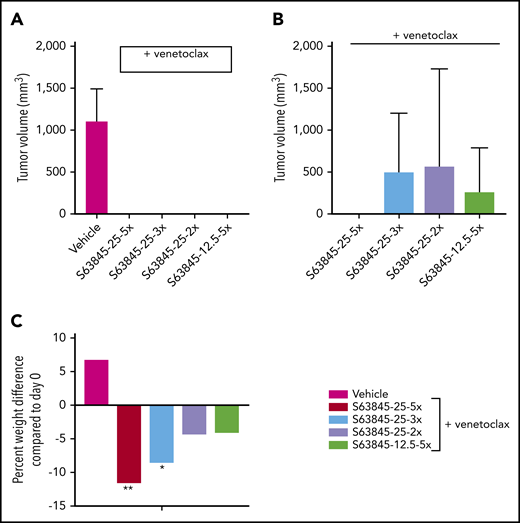
Next, we wanted to investigate whether reduced dosing can still maintain antitumor efficacy. Here, we continued to administer venetoclax at 100 mg/kg orally 5 times per week and reduced the dose of S63845 (Table 1). IV administration of S63845 at 25 mg/kg 2 or 3 times per week in combination with venetoclax for 2 weeks led to complete tumor regression by day 20 of treatment (Figure 4A; supplemental Figure 3A). Tumor regression was possible even by combining venetoclax at 100 mg/kg with S63845 at 12.5 mg/kg IV 5 times per week (Figure 4A; supplemental Figure 3A).
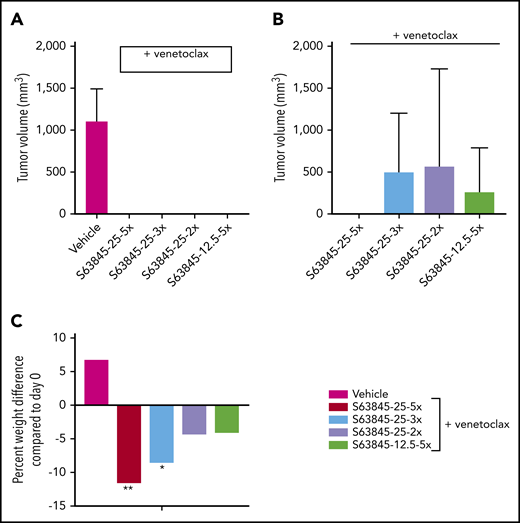
Antitumor efficacy of varying doses of S63845 combined with venetoclax. (A) SU-DHL-6 cells were xenografted subcutaneously into NSG female mice. After 10 days, mice were randomly assigned into comparable groups (5 mice each) and treated with vehicle and combinations of venetoclax (100 mg/kg orally 5 times per week) with varying doses of S63845 (25 mg/kg 2, 3, and 5 times per week and 12.5 mg/kg IV 5 times per week) for 2 weeks. Mean volume and error bars representing SD from 5 different tumors are shown. By day 22 of treatment (day 20 for S63845 at 25 mg/kg 5 times per week condition), there is complete tumor regression in all treatment groups. A 2-way ANOVA was used to determine statistical significance between the first day of treatment and subsequent time points. (B) Tumor volumes in treatment groups from panel A on day 76, tumor regression is maintained only in the group treated with S63845 at 25 mg/kg 5 times per week plus venetoclax at 100 mg/kg 5 times per week. (C) Histogram depicting weight variation in SU-DHL-6 xenografts of different treatment groups on day 26 posttreatment relative to day 0. A 2-way ANOVA was used to determine statistical significance between the first day of treatment and subsequent time points. **P < .01; *P < .05.
Antitumor efficacy of varying doses of S63845 combined with venetoclax. (A) SU-DHL-6 cells were xenografted subcutaneously into NSG female mice. After 10 days, mice were randomly assigned into comparable groups (5 mice each) and treated with vehicle and combinations of venetoclax (100 mg/kg orally 5 times per week) with varying doses of S63845 (25 mg/kg 2, 3, and 5 times per week and 12.5 mg/kg IV 5 times per week) for 2 weeks. Mean volume and error bars representing SD from 5 different tumors are shown. By day 22 of treatment (day 20 for S63845 at 25 mg/kg 5 times per week condition), there is complete tumor regression in all treatment groups. A 2-way ANOVA was used to determine statistical significance between the first day of treatment and subsequent time points. (B) Tumor volumes in treatment groups from panel A on day 76, tumor regression is maintained only in the group treated with S63845 at 25 mg/kg 5 times per week plus venetoclax at 100 mg/kg 5 times per week. (C) Histogram depicting weight variation in SU-DHL-6 xenografts of different treatment groups on day 26 posttreatment relative to day 0. A 2-way ANOVA was used to determine statistical significance between the first day of treatment and subsequent time points. **P < .01; *P < .05.
The reduced dosing regimens induced complete remissions of tumors in mice, but they were not sustained, and tumor regrowth was observed around day 75 (∼50 days after treatment cessation; Figure 4B; supplemental Figure 3A). The onset and growth of the tumor is dependent on the total dose as well as the frequency of administration of S63845 (Table 1). The mice that received S63845 at 50 mg/kg in 1 week (25 mg/kg 2 times per week) were the first to see tumor regrowth, and these tumors grew faster than tumors in other treatment groups. However, mice that received S63845 at 12.5 mg/kg 5 times per week (62.5 mg/kg total per week) developed tumors that grew more slowly than those in mice that received 25 mg/kg 3 times per week (75 mg/kg total per week). Sustained remission was achieved only in the group that was treated with S63845 at 25 mg/kg 5 times per week (125 mg/kg total per week) in combination with venetoclax at 100 mg/kg 5 times per week (Figure 4B). In addition, we continued to observe dose-dependent weight loss (Figure 4C; supplemental Figure 3B). Collectively, these data suggest that the dose of S63845 plus venetoclax required for complete and sustained remission is not well tolerated. To overcome this intolerance, a targeted drug delivery strategy is required. Active targeting of drugs to the tumor can be accomplished with nanoparticles.
Tumor-targeted delivery of S63845 by nanoparticles is efficacious in vivo
To reduce systemic toxicity of S63845 and venetoclax, we used nanoparticle drug carriers to preferentially target P-selectin, an inflammatory cell adhesion molecule that is significantly upregulated in tumor sites, including lymphomas (supplemental Figure 5A).32,33 The nanoparticle formulation of S63845 was designed with 3 key parameters: size, tumor specificity, and ease of visualization. S63845 was first selected for nanoformulation because of its potential toxicity to normal cells when administered systemically.26 The nanoparticles are composed of the small molecule drug S63845, an indocyanine green dye, IR-783, that interacts with the drug and is required for self-assembly into nanoparticles, and fucoidan, a naturally occurring fucosylated polysaccharide with nanomolar affinity to P-selectin (supplemental Figure 4).32-34 Targeting of the drug-loaded nanoparticle to the tumor is achieved by the affinity of fucoidan for P-selectin.32-34 Fucoidan-decorated nanoparticles were synthesized by coencapsulating S63845 and IR-783 (supplemental Figure 4). S63845-encapsulating nanoparticles (S63845-NP) demonstrated an in vitro single-agent activity similar to that of free S63845 (supplemental Figure 5B) and synergized with venetoclax as expected (supplemental Figure 5C).
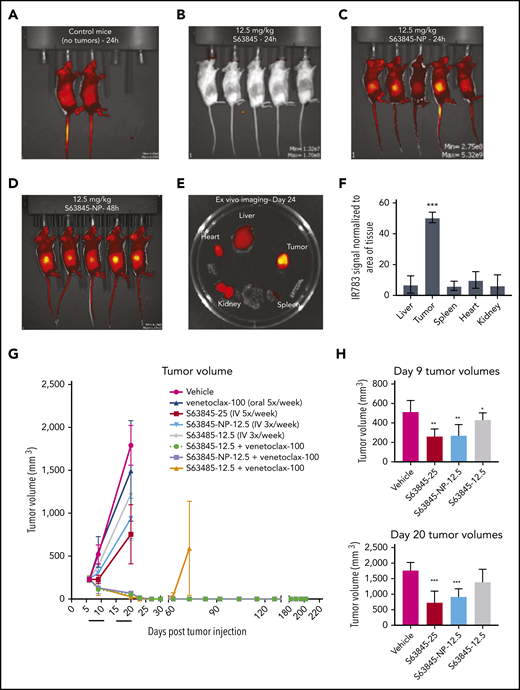
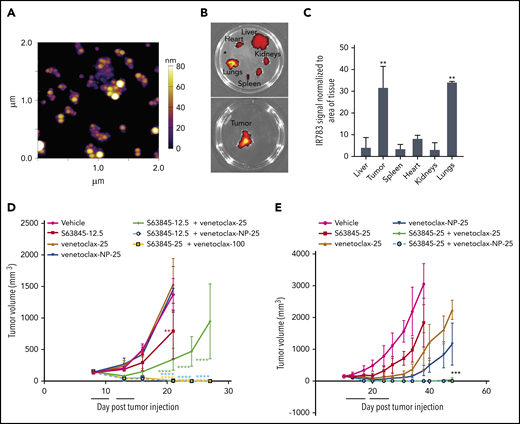
In vivo, the fluorescence intensity of S63845-NP was preferentially localized in the tumors (Figure 5A-E). In control mice that lacked tumors, we observed ubiquitous distribution of S63845-NP (Figure 5A). The biodistribution measured by ex vivo imaging of fluorescence intensity of S63845-NP showed a clear preferential accumulation, ranging from five- to eightfold in the tumor compared with that in other organs (Figure 5E; quantified in Figure 5F).
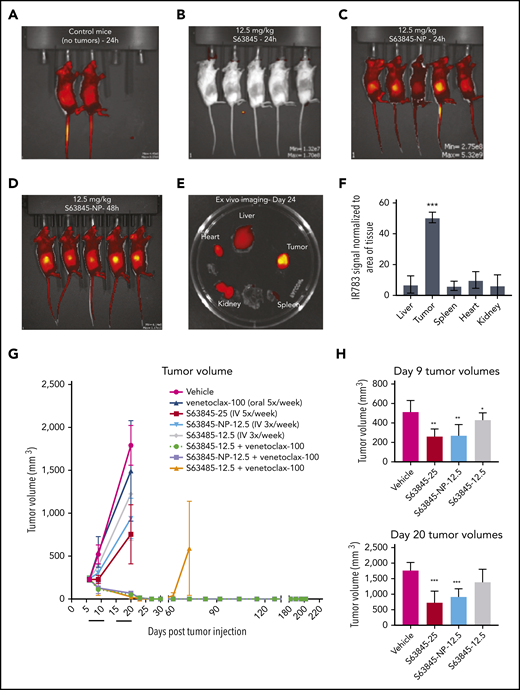
Antitumor efficacy of S63845 as free drug and encapsulated into nanoparticles. (A-E) In vivo fluorescent images acquired via IVIS in vivo imaging technology 24 hours after drug administration. (A) Control mouse treated with S63845-NP. Because these mice lacked tumors, there was ubiquitous distribution of S63845-NP. (B) S63845 was administered but no fluorescence was observed. (C) S63845 nanoparticles were administered. Note the localization of nanoparticles to the tumor. Lack of fur, such as in the ears and tail, also shows high fluorescence. (D) In vivo images 48 hours after administration of S63845 nanoparticles. (E) Ex vivo images of tumor, heart, kidney, spleen, and liver on the day the mice were euthanized (day 24). Nanoparticles were administered 3 times per week for 2 weeks until 24 hours before the day the mice were euthanized (day 24). Note the specific localization of the nanoparticles to the tumor. (F) Nanoparticle biodistribution in organs and tumor, calculated from ex vivo fluorescence images shown in panel E as total fluorescence efficiency divided by tissue area (n = 2) (error bars represent SD). Statistical significance relative to vehicle was calculated by a Mann-Whitney U test. (G) DLBCL cells from the SU-DHL-6 cell line were xenografted subcutaneously into NSG female mice. After 6 days, mice were randomly assigned to comparable groups (4 or 5 mice each) and treated with vehicle, venetoclax (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), S63845 (12.5 mg/kg IV 3 times per week), combinations of venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week) and S63845 (12.5 mg/kg IV 3 times per week) for 2 weeks (indicated by black line). Tumor volumes are represented as means; error bars represent SD. (H) Dot plots comparing tumor sizes on day 9 and 20 after of S63845 single-agent was injected into tumors in various treatment groups: S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week). Tumor volumes are represented as mean ± SD. Statistical significance relative to vehicle was calculated with a 2-way ANOVA. ***P < .001; **P < .01; *P < .05.
Antitumor efficacy of S63845 as free drug and encapsulated into nanoparticles. (A-E) In vivo fluorescent images acquired via IVIS in vivo imaging technology 24 hours after drug administration. (A) Control mouse treated with S63845-NP. Because these mice lacked tumors, there was ubiquitous distribution of S63845-NP. (B) S63845 was administered but no fluorescence was observed. (C) S63845 nanoparticles were administered. Note the localization of nanoparticles to the tumor. Lack of fur, such as in the ears and tail, also shows high fluorescence. (D) In vivo images 48 hours after administration of S63845 nanoparticles. (E) Ex vivo images of tumor, heart, kidney, spleen, and liver on the day the mice were euthanized (day 24). Nanoparticles were administered 3 times per week for 2 weeks until 24 hours before the day the mice were euthanized (day 24). Note the specific localization of the nanoparticles to the tumor. (F) Nanoparticle biodistribution in organs and tumor, calculated from ex vivo fluorescence images shown in panel E as total fluorescence efficiency divided by tissue area (n = 2) (error bars represent SD). Statistical significance relative to vehicle was calculated by a Mann-Whitney U test. (G) DLBCL cells from the SU-DHL-6 cell line were xenografted subcutaneously into NSG female mice. After 6 days, mice were randomly assigned to comparable groups (4 or 5 mice each) and treated with vehicle, venetoclax (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), S63845 (12.5 mg/kg IV 3 times per week), combinations of venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week) and S63845 (12.5 mg/kg IV 3 times per week) for 2 weeks (indicated by black line). Tumor volumes are represented as means; error bars represent SD. (H) Dot plots comparing tumor sizes on day 9 and 20 after of S63845 single-agent was injected into tumors in various treatment groups: S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week). Tumor volumes are represented as mean ± SD. Statistical significance relative to vehicle was calculated with a 2-way ANOVA. ***P < .001; **P < .01; *P < .05.
Next, we compared the efficacy of different doses and schedules of free S63845 and nanoparticle-encapsulated S63845 in vivo. As a single agent, S63845-NP at 12.5 mg/kg administered 3 times per week was more effective than free S63845 administered systemically at the same dose and schedule (27% improvement in tumor regression) (Figure 5H). In addition, S63845-NP at 12.5 mg/kg administered 3 times per week was as effective as S63845 at 25 mg/kg administered 5 times per week, thus allowing us to reduce the administered dose by 3.5-fold (Figure 5H).
When combined with venetoclax administered orally (100 mg/kg 5 times per week), higher doses of both free S63845 (25 mg/kg 5 times per week) and S63845 containing nanoparticles (S63845-NP at 12.5 mg/kg 3 times per week) achieved sustained eradication of xenografted tumors (Figure 5G) and increased survival (supplemental Figure 6A). In contrast, the combination of venetoclax with doses of free S63845 comparable to the nanoparticles (12.5 mg/kg 3 times per week) induced complete remissions that were not durable because tumor regrowth was observed around day 63 (Figure 5G; supplemental Figure 5D-E; doses summarized in Table 2). Collectively, our data demonstrate that encapsulating S63845 into nanoparticles allowed a 3.5-fold reduction of drug administered compared with free S63845.
S63845-NPs measured 268 ± 33 nm in diameter and −55 mV in surface charge (supplemental Figure 5F-H). However, their size increased over time (supplemental Figure 5I). To develop more stable nanoparticles of 50 to 100 nm, which can penetrate the tumor more effectively, we explored encapsulation of venetoclax.
Tumor-targeted delivery of venetoclax by nanoparticles is efficacious in vivo
Recognizing that the encapsulation of venetoclax could also help mitigate toxicities of the combination with S63845, we set out to evaluate the nanoformulation of venetoclax. Venetoclax nanoparticles (venetoclax-NPs) were synthesized using the same parameters and protocol (supplemental Figure 1A). The particles measured 68 ± 7 nm in diameter, had a surface charge of −33 mV, and maintained their size over a 7-day period (Figure 6A; supplemental Figure 7A-D). Free venetoclax and nanoparticle-encapsulated venetoclax demonstrated comparable activity in vitro as a single agent and in combination with S63845 (supplemental Figure 7E-F). In a U-2973 mouse xenograft model, animals treated with venetoclax-NPs showed slower tumor progression than animals treated with comparable doses of free drug (supplemental Figure 7G). In an SU-DHL-6 xenograft model, the antitumor efficacy of venetoclax at 100 mg/kg administered 5 times per week (1000 mg/kg total) and venetoclax at 25 mg/kg administered 3 times per week (150 mg/kg total) was comparable (supplemental Figure 7H). Thus, in this xenograft model, encapsulating venetoclax into nanoparticles enabled a 6.5-fold reduction in the dose of drug administered.
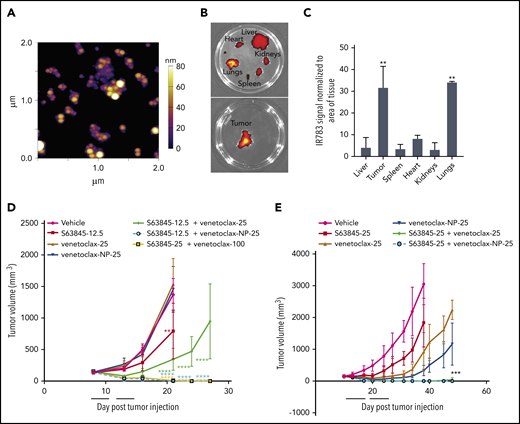
Antitumor efficacy of venetoclax as free drug and encapsulated into nanoparticles. (A) Representative atomic force microscopy images of venetoclax-NP. Size of the nanoparticles is depicted in a colorimetric scale from yellow (80 nm) to dark purple (0 nm). (B) Ex vivo images of tumor (SU-DHL-6), heart, kidney, spleen, and liver on day 14 from a mouse that had venetoclax nanoparticles administered. Note the specific localization of the nanoparticles to tissues (tumor and the lungs) that express P-selectin. (C) Quantification of nanoparticle biodistribution in organs and tumor (SU-DHL-6), calculated from ex vivo fluorescence images shown in panel C as total fluorescence efficiency divided by tissue area (n = 2) (error bars represent SD). Statistical significance relative to vehicle was calculated by a Mann-Whitney U test. (D) DLBCL SU-DHL-6 cells were xenografted subcutaneously into NSG female mice. After 8 days, mice were randomly assigned into comparable groups (5 mice each) and were treated with vehicle, S63845 (12.5 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 3 times per week), venetoclax-NP (25 mg/kg IV 3 times per week), combinations of S63845 (12.5 mg/kg IV 3 times per week) and venetoclax (25 mg/kg orally 3 times per week) and venetoclax-NP (25 mg/kg orally 3 times per week), and venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week) for 2 weeks (indicated by black line). (E) DLBCL U-2973 cells were xenografted subcutaneously into NSG female mice. After 10 days, mice were randomly assigned into comparable groups (5 mice each) and treated with vehicle, S63845 (25 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 5 times per week), IR-783 and venetoclax-NP (25 mg/kg IV 3 times per week), combinations of S63845 (25 mg/kg IV 3 times per week) and venetoclax (25 mg/kg orally 5 times per week), and a combination of IR-783 and venetoclax-NP (25 mg/kg orally 3 times per week) and S63845 (25 mg/kg IV 3 times per week) for 2 weeks (indicated by black line). Tumor volumes are represented as means; error bars represent SD. Statistical difference between tumor group and vehicle at each time point is calculated by 2-way ANOVA. ****P < .0001; ***P < .001; **P < .01.
Antitumor efficacy of venetoclax as free drug and encapsulated into nanoparticles. (A) Representative atomic force microscopy images of venetoclax-NP. Size of the nanoparticles is depicted in a colorimetric scale from yellow (80 nm) to dark purple (0 nm). (B) Ex vivo images of tumor (SU-DHL-6), heart, kidney, spleen, and liver on day 14 from a mouse that had venetoclax nanoparticles administered. Note the specific localization of the nanoparticles to tissues (tumor and the lungs) that express P-selectin. (C) Quantification of nanoparticle biodistribution in organs and tumor (SU-DHL-6), calculated from ex vivo fluorescence images shown in panel C as total fluorescence efficiency divided by tissue area (n = 2) (error bars represent SD). Statistical significance relative to vehicle was calculated by a Mann-Whitney U test. (D) DLBCL SU-DHL-6 cells were xenografted subcutaneously into NSG female mice. After 8 days, mice were randomly assigned into comparable groups (5 mice each) and were treated with vehicle, S63845 (12.5 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 3 times per week), venetoclax-NP (25 mg/kg IV 3 times per week), combinations of S63845 (12.5 mg/kg IV 3 times per week) and venetoclax (25 mg/kg orally 3 times per week) and venetoclax-NP (25 mg/kg orally 3 times per week), and venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week) for 2 weeks (indicated by black line). (E) DLBCL U-2973 cells were xenografted subcutaneously into NSG female mice. After 10 days, mice were randomly assigned into comparable groups (5 mice each) and treated with vehicle, S63845 (25 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 5 times per week), IR-783 and venetoclax-NP (25 mg/kg IV 3 times per week), combinations of S63845 (25 mg/kg IV 3 times per week) and venetoclax (25 mg/kg orally 5 times per week), and a combination of IR-783 and venetoclax-NP (25 mg/kg orally 3 times per week) and S63845 (25 mg/kg IV 3 times per week) for 2 weeks (indicated by black line). Tumor volumes are represented as means; error bars represent SD. Statistical difference between tumor group and vehicle at each time point is calculated by 2-way ANOVA. ****P < .0001; ***P < .001; **P < .01.
In vivo, the fluorescence intensity of venetoclax-NP was highly localized in the tumors (supplemental Figure 9A). The biodistribution measured by ex vivo imaging of fluorescence intensity of venetoclax-NP showed a clear preferential accumulation in the tumor, ranging from threefold to sixfold (Figure 6B, quantified in Figure 6C). Furthermore, mass spectrometry analyses comparing venetoclax concentration upon systemic vs nanoparticle administration revealed a 27% increase in the tumor, 43% decrease in plasma, and a decrease in other organs (supplemental Figure 7I).
Mice treated with a combination of S63845 at 12.5 mg/kg and venetoclax-NP at 25 mg/kg administered 3 times per week showed complete tumor regression within 10 treatments, whereas this was not achieved by the same dosing regimen with free venetoclax. Within the first 10 days of treatment, the combination of venetoclax-NP at 25 mg/kg with S63845 at 12.5 mg/kg showed more efficacious tumor regression than the higher-dose free drug combination of venetoclax at 100 mg/kg 5 times per week plus S63845 at 25 mg/kg 5 times per week. Thus, by encapsulating venetoclax into nanoparticles the efficacious doses of S63845 and venetoclax were successfully reduced 3.5-fold and 6.5-fold, respectively, in the combination strategy (Figure 6D; supplemental Figure 6B; Table 3). These results were confirmed on a second mouse xenograft model using the U-2973 DLBCL cell line (Figure 6E). Mice treated with a combination of S63845 at 25 mg/kg 5 times per week and venetoclax-NP at 25 mg/kg 3 times per week showed complete tumor regression and increased survival in 60% of the mice by day 7 of treatment, similar to that in mice that were administered free agents more frequently (both agents 5 times per week) (Figure 6E; supplemental Figure 6C). However, encapsulation of the 2 drugs was as safe as 1-drug encapsulation, and it maintained similar efficacy toward tumor regression and survival (supplemental Figure 10).
Combination therapy with 1 drug encapsulated in nanoparticles reduces toxicity
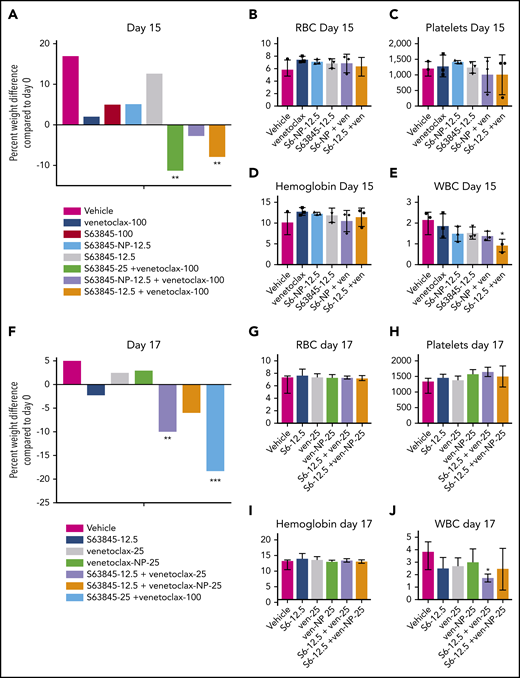
We observed a clear therapeutic advantage in combination therapy when 1 drug was encapsulated into nanoparticles. This was manifested as improved efficacy as previously shown (Figures 5G-H and 6D-E; supplemental Figure 7G-H) along with decreased toxicity. Unlike S63845 at 12.5 mg/kg or 25 mg/kg, S63845-NP at 12.5 mg/kg caused no weight loss in combination with venetoclax (Figure 7A; supplemental Figure 8A-B). Furthermore, at this dose, animals treated with S63845 free drug or S63845-NP plus venetoclax did not display significant deviation from control animals in peripheral blood WBC and RBC counts (Figure 7B-E). Similarly, unlike venetoclax at 25 mg/kg or 100 mg/kg, venetoclax-NP at 25 mg/kg in combination with S63845 did not cause significant weight loss (Figure 7F; supplemental Figure 8C-D). Therapy with a combination of S63845 and venetoclax-NP also did not lead to toxicity in peripheral blood WBCs and RBCs (Figure 7G-J).
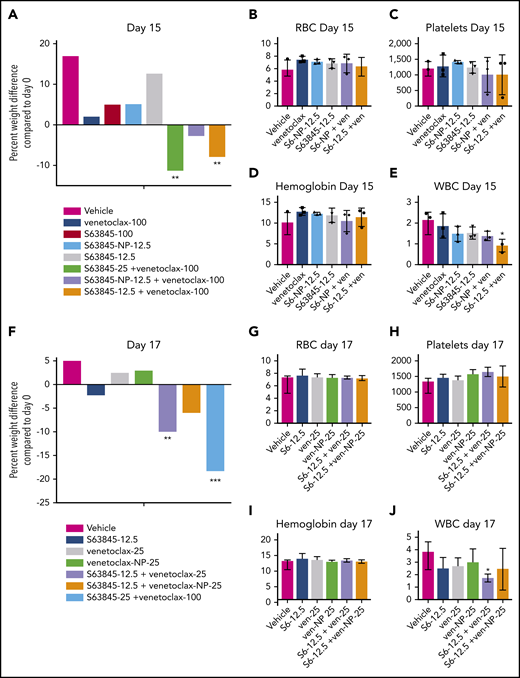
Encapsulation of drug into nanoparticles reduces toxicity associated with combination therapy. (A) Histogram depicting weight variation in SU-DHL-6 xenografts in different treatment groups on day 15 relative to day 0 of treatment: vehicle, venetoclax (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week), combinations of venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week) for 2 weeks (n = 4 or 5 mice). (B-E) Analysis of the effect of combination therapy on RBCs (B), platelets (C), hemoglobin (D), and WBCs (E) on day 15 of treatment. Mean and SD from 5 different mice per group. Treatment groups include vehicle, venetoclax (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week) and combinations of venetoclax (100 mg/kg orally 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week). (F) Histogram depicting weight variation in SU-DHL-6 xenografts in different treatment groups on day 17 relative to day 0 of treatment: vehicle, S63845 (12.5 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 3 times per week), and venetoclax (25 mg/kg IV 3 times per week), combinations of S63845 (12.5 mg/kg IV 3 times per week) and venetoclax (25 mg/kg orally 3 times per week), and venetoclax-NP (25 mg/kg IV 3 times per week), and S63845 (25 mg/kg IV 5 times per week) plus venetoclax (100 mg/kg orally 5 times per week) for 2 weeks. (G-J) Analysis of the effect of combination therapy on RBCs (G), platelets (H), hemoglobin (I), and WBCs (J) on day 15 of treatment. Treatment groups include vehicle, S63845 (12.5 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 3 times per week), and venetoclax (25 mg/kg IV 3 times per week), combinations of S63845 (12.5 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 3 times per week), and venetoclax-NP (25 mg/kg IV 3 times per week). For weight loss, 2-way ANOVA was used and for hematologic toxicity, 1-way ANOVA was used to determine statistical significance between first day of treatment and subsequent time points. ***P < .001; **P < .01; *P < .05.
Encapsulation of drug into nanoparticles reduces toxicity associated with combination therapy. (A) Histogram depicting weight variation in SU-DHL-6 xenografts in different treatment groups on day 15 relative to day 0 of treatment: vehicle, venetoclax (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week), combinations of venetoclax (100 mg/kg orally 5 times per week) and S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week) for 2 weeks (n = 4 or 5 mice). (B-E) Analysis of the effect of combination therapy on RBCs (B), platelets (C), hemoglobin (D), and WBCs (E) on day 15 of treatment. Mean and SD from 5 different mice per group. Treatment groups include vehicle, venetoclax (100 mg/kg orally 5 times per week), S63845 (25 mg/kg IV 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week) and combinations of venetoclax (100 mg/kg orally 5 times per week), S63845-NP (12.5 mg/kg IV 3 times per week), and S63845 (12.5 mg/kg IV 3 times per week). (F) Histogram depicting weight variation in SU-DHL-6 xenografts in different treatment groups on day 17 relative to day 0 of treatment: vehicle, S63845 (12.5 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 3 times per week), and venetoclax (25 mg/kg IV 3 times per week), combinations of S63845 (12.5 mg/kg IV 3 times per week) and venetoclax (25 mg/kg orally 3 times per week), and venetoclax-NP (25 mg/kg IV 3 times per week), and S63845 (25 mg/kg IV 5 times per week) plus venetoclax (100 mg/kg orally 5 times per week) for 2 weeks. (G-J) Analysis of the effect of combination therapy on RBCs (G), platelets (H), hemoglobin (I), and WBCs (J) on day 15 of treatment. Treatment groups include vehicle, S63845 (12.5 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 3 times per week), and venetoclax (25 mg/kg IV 3 times per week), combinations of S63845 (12.5 mg/kg IV 3 times per week), venetoclax (25 mg/kg orally 3 times per week), and venetoclax-NP (25 mg/kg IV 3 times per week). For weight loss, 2-way ANOVA was used and for hematologic toxicity, 1-way ANOVA was used to determine statistical significance between first day of treatment and subsequent time points. ***P < .001; **P < .01; *P < .05.
Targeted tumor delivery using nanoparticles allows a reduction in drug dose and thus a reduction in the associated toxicity. Note that with both S63845 and venetoclax, lowering the dose of these drugs combined and administered systemically led to decreased toxicity and efficacy, whereas nanoparticle encapsulation decreased toxicity but increased the efficacy of the drugs. Thus, we demonstrated a clear therapeutic benefit of encapsulating S63845 or venetoclax into nanoparticles for use in combination therapy.
Discussion
In this study, we demonstrated the efficacy of the recently described MCL1 inhibitor S63845 across a range of lymphoma subtypes. We and others previously reported that MCL1 may compensate for BCL2 inhibition in lymphoma, suggesting the need for dual inhibition of MCL1 and BCL2 to enhance the efficacy of venetoclax.7,20 Consistent with this hypothesis, we demonstrated that this combination was highly effective in vivo, because only 9 treatments were sufficient to induce durable remissions in a DLBCL xenograft model.
MCL1 is essential for the survival of hematopoietic stem cells,35 and early and late development and survival of B and T lymphocytes.36 MCL1 knockout mice will show peri-implantation embryonic lethality.22 MCL1 inhibition has been shown to induce cardiomyocyte toxicity.26 Furthermore, preclinical analyses of MCL1 inhibition are confounded because S63845 has a sixfold higher affinity for human MCL1 relative to mouse MCL1.10 Thus there are significant barriers to safety when introducing an MCL1 inhibitor to the clinic, which is underscored by a recent clinical hold on a phase 1 dose escalation study of AMG 397.37 Given the essential roles of MCL1 and BCL2 on the survival of hematopoietic cells and other normal tissues, there is a concern that dual inhibition of both proteins would be associated with intolerable toxicity in patients with cancer.38 Indeed, we found that the combination of S63845 and venetoclax was associated with significant hematologic toxicity, which included reduction in circulating WBCs, RBCs, and platelets in addition to rapid and significant weight loss. Using different lower doses of S63845 combined with venetoclax decreased toxicity to the animals, but that was also associated with reduced efficacy because tumor regrowth was observed within 2 months.
An alternative strategy is to use targeted delivery methods (eg, targeted nanoparticles) that preferentially deliver toxic drugs to the tumors while reducing systemic exposure. Here, we encapsulated either S63845 or venetoclax into nanoparticles composed of the polysaccharide fucoidan. We showed that the enhanced therapeutic efficacy of S63845 plus venetoclax-NP was associated with selective accumulation of the nanoparticles in the tumor microenvironment as can be seen by in vivo and ex vivo imaging facilitated by IR-783. We observed up to a sixfold increased fluorescence in tumor sites compared with that in normal tissue. This targeted delivery allowed us to achieve an antitumor efficacy similar to that with systemically administered drug while using 3.5-fold lower doses of S63845 and 6.5-fold lower doses of venetoclax. Encapsulating one drug in nanoparticles while giving the second drug by systemic administration reduced the hematologic toxicity associated with the systemic administration of both drugs, which confirmed that we are achieving preferential drug delivery to the tumor. The reduction of toxicity was observed by encapsulation of either S63845 or venetoclax into nanoparticles. The sixfold higher affinity of S63845 to human MCL110 would increase the potential toxicity of S63845 as a single agent and in combination with venetoclax in humans or in a humanized MCL1 mouse model. Thus, quantifying how encapsulation into nanoparticles decreases toxicity in such a model would be informative.
P-selectin–targeted nanoparticles have been used to encapsulate drugs targeting other types of cancers,32,39-41 but our study is the first to use this strategy to selectively deliver BH3 mimetics to lymphoma sites. To apply this strategy in a clinical setting, several technical aspects will need to be addressed, especially the ability to produce a uniform encapsulation of the drugs into nanoparticles. This is essential because the properties of the particles may change significantly by modest changes in size, charge, and other physiochemical properties. With our methods, venetoclax encapsulation resulted in more uniform small size nanoparticles (68 ± 7 nm) compared with S63845-NP (268 ± 33 nm). Future studies should examine new methods for producing nanoparticles consistent in size and drug loading by using technologies such as nanoprecipitation with hydrodynamic flow and microfluidic platforms.42,43 An alternative delivery strategy would be to explore conjugation of the BH3 mimetics to antibodies that selectively target tumor-associated surface proteins.
We anticipate that encapsulation of BH3 mimetics into nanoparticles will allow the simultaneous delivery of additional inhibitors of BCL2 protein family members, such as BCL-xL, which would open the door for several combinations that would be too toxic if given by systemic administration. Furthermore, our targeted delivery approach allows us to revisit drugs such as navitoclax (that targets BCL2 and BCL-xL); even though it showed effectiveness, it was considered too toxic.44
To summarize, in this study we showed the potent antitumor efficacy of MCL1 and BCL2 dual inhibition using S63845 and venetoclax in DLBCL, which can lead to durable remission after only 9 treatments. Furthermore, we were able to significantly ameliorate the toxicity associated with this combination therapy by encapsulating each drug into tumor-targeting nanoparticles. Further development of this strategy for clinical application holds great potential.
For original data, please contact Anas Younes at anas.younes@astrazeneca.com.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge all members of the Younes laboratory for feedback regarding experimental procedures.
This work was supported in part by grants from the National Institutes of Health (NIH)/National Cancer Institute Memorial Sloan Kettering Specialized Program of Research Excellence in Lymphoma (P50 CA192937), Institutional Core Grant (P30 CA008748), grant R01-CA215719-02, and Cancer Center Support Grant (P30 CA008748); and NIH/Eunice Kennedy Shriver National Institute of Child Health and Human Development New Innovator Award (DP2-HD075698). Additional support was provided by The George L. Ohrstrom Jr Foundation Fund, the Leukemia & Lymphoma Society Specialized Center of Research Program (7014-17), the National Science Foundation CAREER Award (1752506), the American Cancer Society Research Scholar Grant (GC230452), the Pershing Square Sohn Cancer Research Alliance, the Expect Miracles Foundation-Financial Services Against Cancer, the Cycle for Survival’s Equinox Innovation Award, and the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center.
Authorship
Contribution: N.B.T. and A.Y. conceived the study; N.B.T., M.T.M., H.K., D.A.H., and A.Y. determined the methodology; V.S. provided the software: N.B.T., L.H., and M.T.M. conducted the formal data analysis; N.B.T., L.H., M.T.M., and M.D.S.F. conducted the investigation; C.H. and E.d.S. managed the animals needed for the study; E.d.S. and K.M.-T. provided resources for the study; N.B.T. and A.Y. wrote the original draft of the manuscript; N.B.T. and A.Y. conducted the visualization aspects of the study; L.H. conducted rebuttal experiments and revision of the manuscript; A.Y. and D.A.H. supervised the study; and D.A.H. and A.Y. acquired the study funding.
Conflict-of-interest disclosure: M.T.M. holds shares in SesenBio and served as a consultant for Synthis. D.A.H. is a cofounder and officer with equity interest in Goldilocks Therapeutics, LipidSense, and Nirova Biosense and served on the scientific advisory boards of Concarlo Holdings and Nanorobotics. A.Y. received research support from Janssen, Curis, Merck, Bristol Myers Squibb, Syndax, and Roche; received honoraria from Janssen, AbbVie, Merck, Curis, Epizyme, Roche, and Takeda; and served as a consultant for Biopath, Xynomics, Epizyme, Roche, Celgene, and HCM. The remaining authors declare no competing financial interests.
The current affiliation for A.Y. is AstraZeneca, Gaithersburg, MD.
Correspondence: Anas Younes, AstraZeneca, One Medimmune Way, Gaithersburg, MD 20878; e-mail: anas.younes@astrazeneca.com.

![S63845 has antiproliferative activity in lymphoma cells and synergizes with venetoclax. (A) Heat map summarizing the IC50 (mean ± standard error of the mean [SEM]) in µM of S63845 and venetoclax in 14 lymphoma cell lines with the following subtypes: activated B-cell (ABC) DLBCL, germinal center B-cell (GCB) DLBCL, double-hit lymphoma (DHL), and mantle cell lymphoma (MCL). Cells were incubated with increasing concentrations of drug for 72 hours. Data represent the mean of 3 separate experiments. Cell viability was assessed by CellTiter-Glo. Sensitive, IC50 <0.1 µM; moderately sensitive, IC50 between 0.1 and 1 µM; resistant, IC50 >1 µM. (B) Dot plots quantifying MCL1, BCL2, BCL-xL, and BIM protein expression from western blot in supplemental Figure 2A in the 4 cell lines most resistant (Res) (U-2932, HBL-1, Ri-1, and Maver-1) and sensitive (Sen) (U-2973, OCI-LY19, Mino, and SU-DHL-6) to S63845. Each point represents the protein:β-actin ratio in a cell line. Error bars represent SD from 4 different cell lines listed above. Statistical significance was measured by an unpaired Student t test. (C) Heat map summarizing the effect of combinations of varying concentrations of S63845 and venetoclax on cell viability in the indicated cell lines as assessed by the CellTiter-Glo assay. Combination responses are examined in a 5 × 5 viability matrix after 24 hours of treatment. Percentage of cell viability is depicted in a colorimetric scale from black (high) to white (low) normalized to dimethyl sulfoxide control. Values are the mean of 3 separate experiments. (D) Box plot graph summarizing the results of treatment with combined S63845 and venetoclax for 24 hours in the 3 cell lines most resistant to S63845 (Ri-1, HBL-1, and U-2932) and venetoclax (TMD8, Z-138, and Jeko-1). The y-axis depicts the log-odds that indicate the ratio between observed and expected inhibition on a log scale. Log-odds >0 signify antagonism, log-odds of 0 signify an additive effect, and log-odds <0 (area below gray line) indicate synergy between the 2 drugs. Cell viability was assessed by CellTiter-Glo. All data points represent the mean of 3 separate experiments. ***P < .001; **P < .01; *P = .02.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/15/10.1182_blood.2020008017/2/m_bloodbld2020008017f1.png?Expires=1769402103&Signature=rXnc~2D~7xy9XX6xc4jyz~4yhNrSXyp9oDku0v8IEVoQ39GqjPZCF0Tx5NDSjt5x1wpA6cHdxBdjw73Tfji7MY6Xu3u2fvZ2NFkk1OIcrkeWovqFUfYzvkXqoymPH~0CKp4rwOsH2cabXioq1GlTdDZTa5ehV7PqDMfzrYyb-ev0SBDu2quh5PF~wJ7qMUQx8FqXPjbfBgSjzvfLrD~FPbRYlD4CmEcpQbccJtWohE6t3Yn3AhFLjzO27vXe7LIZLZ4f9Q7cye~0O41f7rimYh-Q1WvIe3EeyG2NmPyhO2VXUBhw~vVut7ZtKsBgEbK92UdLNA5m6BlM9jhplSsC9Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)