Key Points
CRISPR/Cas9 repair of CYBB mutations with an oligodeoxynucleotide restores endogenous regulation of expression in X-CGD CD34+ cells.
Gene-editing enhancers greatly increase homology-directed repair to achieve highly efficient gene correction of engrafting CD34+ cells.

Abstract
Lentivector gene therapy for X-linked chronic granulomatous disease (X-CGD) has proven to be a viable approach, but random vector integration and subnormal protein production from exogenous promoters in transduced cells remain concerning for long-term safety and efficacy. A previous genome editing–based approach using Streptococcus pyogenes Cas9 mRNA and an oligodeoxynucleotide donor to repair genetic mutations showed the capability to restore physiological protein expression but lacked sufficient efficiency in quiescent CD34+ hematopoietic cells for clinical translation. Here, we report that transient inhibition of p53-binding protein 1 (53BP1) significantly increased (2.3-fold) long-term homology-directed repair to achieve highly efficient (80% gp91phox+ cells compared with healthy donor control subjects) long-term correction of X-CGD CD34+ cells.
Introduction
Chronic granulomatous disease (CGD) is a genetic immune deficiency caused by defects in the phagocyte NADPH oxidase (NOX2), a multicomponent enzyme complex that generates microbicidal reactive oxidative species (ROS).1 Defects in gp91phox, the main catalytic subunit of NOX2, cause the X-linked form of CGD (X-CGD) that accounts for 70% of CGD cases. Defects in any 1 of 4 other subunits (p47phox, p22phox, p67phox, and p40phox) cause autosomal forms of CGD.
Despite significant advances in antimicrobial therapy, infections remain the biggest cause of early mortality.2 For patients, residual ROS production remains a prognostic determinant of survival regardless of the variable genetic mutations that cause CGD.3 Hence, therapeutic efforts to improve residual ROS production in phagocytes are contingent on promoting sufficient NOX2 protein production to support near-normal cellular ROS generation. Consequently, a challenge for the use of lentiviral vector–mediated gene transfer4 or safe harbor–targeted gene insertion5 for gene therapy of CGD has been achieving normal physiological levels of NOX2 expression and ROS production in corrected cells using exogenous promoters to regulate gene expression.
We previously showed that CRISPR-Cas9/oligodeoxynucleotide (ODN)–mediated gene repair of a CYBB exon 7 c.676 C>T mutation in X-CGD patients’ CD34+ hematopoietic stem/progenitor cells (HSPCs) restored regulation of CYBB expression by the endogenous promoter and achieved normal per-cell levels of ROS production in HSPC-derived neutrophils.6 The CRISPR/Cas9–induced double-strand DNA break (DSB) was repaired by homology-directed repair (HDR), which resulted in a conversion of the CYBB mutation to the desired functional sequence provided by a short 100-nucleotide ODN donor template. Unfortunately, the long-term gene correction rates in that study were typically <10% posttransplantation, which severely hampered the prospect of clinical translation. HDR activity is a limiting factor for the repair process in quiescent CD34+ HSPCs that is restricted to the G2 and S phases of the cell cycle.7 Herein we sought to optimize HDR of a corrective donor template following Cas9/single guide RNA (sgRNA)-induced DSB to address the variable and inadequate gene correction levels in long-term engrafting CD34+ HSPCs necessary for clinical translation.
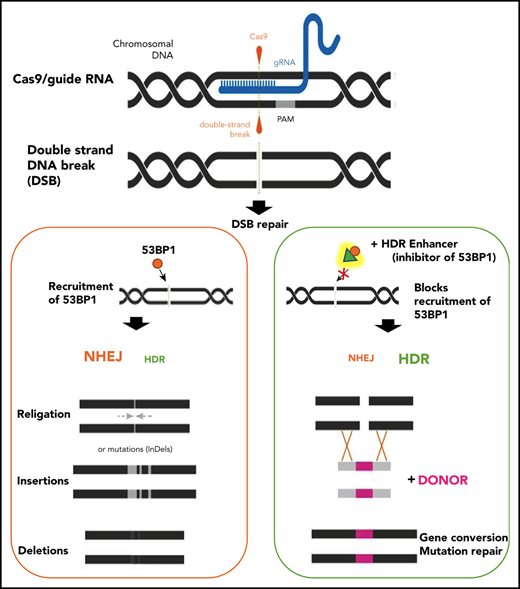
A key regulator of HDR activity is the p53-binding protein 1 (53BP1), a large multidomain chromatin-binding protein and a master regulator of DSB repair pathway choice. 53BP1 greatly favors nonhomologous end-joining (NHEJ) by suppressing end resections that are necessary for HDR. Not surprisingly, there has been much interest in the manipulations of 53BP1.8,9 The recruitment and accumulation of 53BP1 at DSB sites are dependent on its tandem Tudor domain (TTD) interacting with ubiquitylated histone H2A. i53 is an engineered ubiquitin variant with a high binding affinity to the TTD of 53BP1, and it effectively blocks 53BP1 recruitment to damaged chromatin through occlusion of the TTD ligand-binding site. The addition of i53 therefore interferes with 53BP1 accumulation at DSBs, effectively reduces NHEJ repair, and promotes HDR, as shown previously in mammalian cell lines, or as a fusion protein with Cas9.8,10
Materials and methods
Human blood cells
Human CD34+ HSPCs were collected from healthy male donors or male patients with X-CGD after obtaining written informed consent (National Institute of Allergy and Infectious Diseases protocols 05-I-0213 and 94-I-0073).
Editing reagents
All Cas9 messenger RNAs (mRNAs) were provided by CELLSCRIPT, LLC (supplemental Material, available on the Blood Web site), except for chemically modified synthetic guides (Synthego).11 Chemically modified single-strand ODN donor or adeno-associated vectors (AAVs; Vigene Biosciences) were used for correction studies.12
Targeted genome editing in CD34+ HSPCs
Cryopreserved CD34+ HSPCs were prestimulated for 2 days before delivery of gene editing (GE) reagents by electroporation (HPSC34-3 protocol [MaxCyte] or 4D-Nucleofector System with program DZ-100 [Lonza]) (supplemental Material).
Molecular analysis of targeted editing in HSPCs
Genomic DNA was extracted at 2 to 5 days post-electroporation for evaluation of cutting and correction efficiencies and off-target (OT) analysis. Targeted deep sequencing quantified allele repairs and insertions and deletions (indels) (Table 1). Long-range polymerase chain reaction (PCR) was performed by using nested PCRs and sequenced by using PacBio.
In vitro myeloid differentiation
CD34+ HSPCs underwent myeloid differentiation for analyses of mature phagocyte gp91phox expression and NADPH oxidase function6 (supplemental Material).
Transplantation of CD34+ HSPCs into NSG mice
CD34+ cells were transplanted into 6- to 8-week old immunodeficient NOD.SCIDy-Prkdcscid Il2rgtm1Wjl/SzJ mice (The Jackson Laboratory, JAX5557) (National Institute of Allergy and Infectious Diseases Institutional ACUC Protocol LCIM-1E) (supplemental Material).
Karyotyping and fluorescent in situ hybridization
Statistical analysis
The nonparametric Mann-Whitney U test was used for statistical analyses of targeted insertion efficiency (from droplet digital PCR analysis and green fluorescent protein [GFP] or gp91phox expression analyses) (GraphPad Software). For comparing the on-target and OT indel rates, an ordinary one-way analysis of variance test was applied.
Results
Normal-level ROS per cell more protective than high percentages of low-level ROS+ cells
Phagocytic ROS production in vitro can be detected by using a flow cytometry–based dihydrorhodamine 123 (DHR) assay to evaluate ROS production in individual cells following stimulation, which oxidizes DHR to fluorescent rhodamine. An alternative is quantitative ROS measurement by O2– reduction of ferricytochrome c following stimulation yielding an average response of pooled (responding and nonresponding) cells.15 To determine whether per-cell ROS or pooled net ROS production was prognostic for CGD, we compared DHRs of low protein per cell in male X-CGD patients (n = 128) vs female X-CGD carriers (n = 187). These carriers have heterogeneous cell populations with a mixture of normal functioning (DHR+) cells in which the X-chromosome allele carrying the mutated CYBB gene is inactivated, and abnormal functioning cells (DHR–) in which the X-chromosome allele carrying the normal sequence CYBB gene is instead inactivated (Figure 1A). Importantly, we found that although most X-CGD carriers with ∼20% normal functioning myeloid cells (∼45-50 nmoles of O2–) are asymptomatic, the low level of ROS produced per cell in some X-CGD patients (primarily patients with missense mutations)3 seems insufficient to protect them from infections despite the fact they exhibit comparable levels of net O2– production. The mean level of superoxide production compared with that of healthy donor (HD) volunteers (n = 1095) is lower in X-CGD carriers (n = 187) overall but is at the same level if normalized to the percentages of DHR+ cells (ie, amount of ROS produced per DHR+ cell). Patients with X-CGD (n = 128) produced very low amounts of superoxide (Figure 1B).
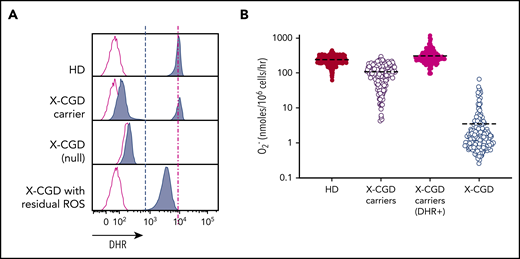
DHR assay and quantitative assessment of superoxide production highlights X-CGD carriers with reduced-frequency functioning phagocytes to produce normal level O2– per cell. (A) FACS DHR assay comparing ROS (blue) production in HD subjects, X-CGD carriers, and X-CGD patients with null mutation, or missense low residual ROS producing, compared with unstimulated controls (red). (B) Quantitative superoxide measurement in pooled cells from HD subjects (n = 1095), X-CGD carriers (n = 187) (pooled cells or normalized to DHR+ cells), and X-CGD patients (n = 128).
DHR assay and quantitative assessment of superoxide production highlights X-CGD carriers with reduced-frequency functioning phagocytes to produce normal level O2– per cell. (A) FACS DHR assay comparing ROS (blue) production in HD subjects, X-CGD carriers, and X-CGD patients with null mutation, or missense low residual ROS producing, compared with unstimulated controls (red). (B) Quantitative superoxide measurement in pooled cells from HD subjects (n = 1095), X-CGD carriers (n = 187) (pooled cells or normalized to DHR+ cells), and X-CGD patients (n = 128).
Optimization of HDR in CD34+ HSPCs
To improve long-term gene correction after genome editing of X-CGD patient CD34+ HSPCs,6 we sought to achieve functional CYBB gene correction at the exon 7 locus by optimizing 2 main CRISPR-based approaches: (1) repair of the CYBB exon 7 c.676 C>T mutation using a 100-nucleotide ODN donor; or (2) targeted insertion (TI) of a relatively large (∼2.1 kb, including homology arms) donor encoding CYBB exon 7 to 13 complementary DNA (cDNA) (CYBB E7-13pA, or CYBB E7-13pA woodchuck hepatitis virus posttranscriptional regulatory element [WPRE]), delivered by an AAV.12 The latter TI approach should address all CYBB mutations that occur at and downstream of CYBB exon 7, encompassing >50% of X-CGD patients.16 A CYBB E7-13 donor (CYBB E7-13pA-SFFV GFP) that coexpresses GFP was designed to allow use of CD34+ HSPCs from HDs for genome editing optimization (Figure 2A). The sgRNA for mutation repair (sg7) targets the CYBB c.676 C>T mutation specifically, whereas the sgRNA for TI (sg1) targets the normal gene sequence at the same locus.
![HDR enhancement with 53BP1 inhibition is most effective with i53 mRNA. (A) Depiction of CYBB c.676 C>T mutation (top; red arrowhead) and successful repair with a 100-nucleotide ODN donor or TI of CYBB E7-13pA, CYBB E7-13pA-WPRE, or CYBB E7-13pA-SFFV-GFP delivered by AAV. (B) Percentages of GFP+ in HD HSPCs following TI with increasing i53 concentration (0-300 µg/mL); (n = 1; 8 replicates). (C) Percentages of GFP+ cells GE with Cas9 RNA or RNP and i53 or e18 enhancer (n = 4 experiments; 2 donors). (D) TI rates after GE with Cas9 RNA or RNP and increasing AAV donor (multiplicity of infection 1 = 103/cell) ±i53 (150 µg/mL; n = 3 [1 HD, 2 X-CGD patients]). (E) TI rates after GE in the presence of i53, BRCA2, or RAD51 at indicated concentrations (n = 2 experiments). *P < .05, **P < .01, ****P < .0001, paired Student t test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/19/10.1182_blood.2020008503/1/m_bloodbld2020008503f2.png?Expires=1768894606&Signature=JQUgwqbi3ePTYzmYpluazjx~dIN95G-BLT~rGyX~yAglMNBV2IZct69mb0sky4vkgtOlKHk4lhfyxm9gMR6F9WV0rLad8kDnUhVABl4N6DNpkZFTNBzfFH6kN29lwwPqk16sHlSfIL~ykbz6UPo88T8Whr-sNXvrN9KTNpKM8QP3LEjHj1DsaqqIoeDHOYc248cmiNUPI4tq~oZg43au00QPkD7tCQrso0zNcqw4ZZNuKQXWiIuEAhamvaim~GFZlDVTQFvDwVV1NdxdbxHORjZsHsQTeNq4jI7YL30X6Sm~1FHqbugj6neuYhxNMqXkaSg54XWz22yYSRdg-PQndQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HDR enhancement with 53BP1 inhibition is most effective with i53 mRNA. (A) Depiction of CYBB c.676 C>T mutation (top; red arrowhead) and successful repair with a 100-nucleotide ODN donor or TI of CYBB E7-13pA, CYBB E7-13pA-WPRE, or CYBB E7-13pA-SFFV-GFP delivered by AAV. (B) Percentages of GFP+ in HD HSPCs following TI with increasing i53 concentration (0-300 µg/mL); (n = 1; 8 replicates). (C) Percentages of GFP+ cells GE with Cas9 RNA or RNP and i53 or e18 enhancer (n = 4 experiments; 2 donors). (D) TI rates after GE with Cas9 RNA or RNP and increasing AAV donor (multiplicity of infection 1 = 103/cell) ±i53 (150 µg/mL; n = 3 [1 HD, 2 X-CGD patients]). (E) TI rates after GE in the presence of i53, BRCA2, or RAD51 at indicated concentrations (n = 2 experiments). *P < .05, **P < .01, ****P < .0001, paired Student t test.
HDR enhancement with 53BP1 inhibition is most effective with i53 mRNA. (A) Depiction of CYBB c.676 C>T mutation (top; red arrowhead) and successful repair with a 100-nucleotide ODN donor or TI of CYBB E7-13pA, CYBB E7-13pA-WPRE, or CYBB E7-13pA-SFFV-GFP delivered by AAV. (B) Percentages of GFP+ in HD HSPCs following TI with increasing i53 concentration (0-300 µg/mL); (n = 1; 8 replicates). (C) Percentages of GFP+ cells GE with Cas9 RNA or RNP and i53 or e18 enhancer (n = 4 experiments; 2 donors). (D) TI rates after GE with Cas9 RNA or RNP and increasing AAV donor (multiplicity of infection 1 = 103/cell) ±i53 (150 µg/mL; n = 3 [1 HD, 2 X-CGD patients]). (E) TI rates after GE in the presence of i53, BRCA2, or RAD51 at indicated concentrations (n = 2 experiments). *P < .05, **P < .01, ****P < .0001, paired Student t test.
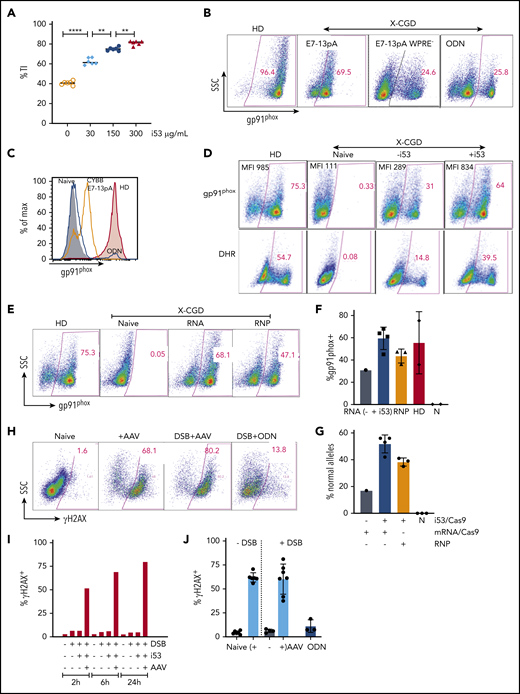
First, we assessed agents with reported boosting effects on efficiency of gene insertion in cell lines, induced pluripotent stem cells, or mouse oocytes8 that purportedly increase HDR directly, or indirectly increase HDR by suppressing NHEJ via inhibition of 53BP1. We first evaluated the impact of i53, a protein-based 53BP1 inhibitor. Genome editing of granulocyte-colony stimulating factor–mobilized peripheral blood CD34+ HSPCs from HDs was performed with Cas9 mRNA/sgRNA and AAV delivered donor (CYBB E7-13pA-SFFV GFP) to facilitate easy detection of TI with GFP expression (Figure 2B). We observed increased percentages of GFP+ cells with increasing i53 mRNA concentration (maximum at 300 µg/mL) to achieve 66.8% GFP+ cells (mean) vs 29.6% (mean) for control without i53 (n = 1 [8 replicates]). We next compared the effect of i53 mRNA vs another protein-based 53BP1 inhibitor, an engineered RAD18 variant (e18),9 when edited with Cas9 mRNA (RNA) or as nuclease (ribonucleoprotein [RNP]) (n = 4 experiments; 2 donors) (Figure 2C). TI efficiency was substantially higher in cells treated with i53, especially when edited with Cas9 RNA (n = 4 experiments, 2 donors). Next, we also observed i53-enhanced TI of AAV-CYBB E7-13pA WPRE when edited with Cas9 RNA or RNP platforms at different AAV donor amounts (n = 3 experiments [2 X-CGD donors, 1 healthy donor]; AAV 1 represents 103/cell) (Figure 2D).
Because HDR activity is dependent on other endogenous HDR pathway proteins such as BRCA217 and RAD52,17,18 we sought to determine if cotransfecting the cells at the time of genome editing electroporation with mRNAs encoding for BRCA2 or RAD52 would also improve HDR-mediated TI. However, neither performed as well as i53 and, when combined, negatively affected the TI percentage compared with i53 alone (Figure 2E).
TI of AAV-CYBB exon 7 to 13 cDNA in X-CGD CD34+ HSPCs in vitro
We next evaluated whether TI of CYBB E7-13 cDNA could functionally correct mutations downstream of E7. We applied Cas9/sgRNA and AAV-CYBB E7-13pA donor to a X-CGD patient (mutation in exon 12) CD34+ HSPCs, and observed a similar TI rate that doubled with increasing i53 concentrations (0 to 300 µg/mL) as measured by droplet digital PCR using donor-locus specific primer pairs (Figure 3A).
Optimization of HDR-dependent GE conditions to functionally correct X-CGD HSPCs and DNA damage response triggered by AAV/DSB. (A) TI CYBB E7-13pA in X-CGD CD34+ HSPCs with i53 as indicated. (B) Dot plots of gp91phox expression in myeloid-differentiated HSPCs gene edited with AAV (E7-13pA and E7-13pA WPRE) or ODN. (C) FACS histograms comparing gp91phox expression in X-CGD E7 mutation repair by ODN (navy line) aligned with HD (shaded red) and AAV-TI CYBB E7-13pA (orange line), compared with naive control (gray). (D) Representative FACS of gp91phox expression (top) and DHR assay (bottom) in myeloid-differentiated X-CGD CD34+ cells after CYBB E7 ODN mutation repair ±i53 (150 µg/mL). (E) FACS comparison of gp91phox expression in differentiated X-CGD HSPCs GE with Cas9 RNA or RNP. (F) Percentages of gp91phox expressing cells with ODN mutation repair of X-CGD HSPCs using Cas9 mRNA ±i53 or RNP +i53 (150 µg/mL). (G) Sequencing shows repaired alleles in HSPCs GE with Cas9 RNA or RNP and ODN ±i53 (150 µg/mL). (H) FACS for phosphorylated H2AX (γH2AX) in HSPCs exposed to AAV alone, or with DSB repaired with AAV or ODN donor at 2 hours. (I) The kinetics of γH2AX expression in HSPCs exposed to AAV with or without DSB and i53 (150 µg/mL). (J) Phosphorylation of H2AX after exposure of HSPCs to AAV (4 different donors) alone or with DSB repair by AAV or ODN. n = 4 experiments. **P < .01, ****P < .0001, paired Student t test. N, naive; SSC, side scatter.
Optimization of HDR-dependent GE conditions to functionally correct X-CGD HSPCs and DNA damage response triggered by AAV/DSB. (A) TI CYBB E7-13pA in X-CGD CD34+ HSPCs with i53 as indicated. (B) Dot plots of gp91phox expression in myeloid-differentiated HSPCs gene edited with AAV (E7-13pA and E7-13pA WPRE) or ODN. (C) FACS histograms comparing gp91phox expression in X-CGD E7 mutation repair by ODN (navy line) aligned with HD (shaded red) and AAV-TI CYBB E7-13pA (orange line), compared with naive control (gray). (D) Representative FACS of gp91phox expression (top) and DHR assay (bottom) in myeloid-differentiated X-CGD CD34+ cells after CYBB E7 ODN mutation repair ±i53 (150 µg/mL). (E) FACS comparison of gp91phox expression in differentiated X-CGD HSPCs GE with Cas9 RNA or RNP. (F) Percentages of gp91phox expressing cells with ODN mutation repair of X-CGD HSPCs using Cas9 mRNA ±i53 or RNP +i53 (150 µg/mL). (G) Sequencing shows repaired alleles in HSPCs GE with Cas9 RNA or RNP and ODN ±i53 (150 µg/mL). (H) FACS for phosphorylated H2AX (γH2AX) in HSPCs exposed to AAV alone, or with DSB repaired with AAV or ODN donor at 2 hours. (I) The kinetics of γH2AX expression in HSPCs exposed to AAV with or without DSB and i53 (150 µg/mL). (J) Phosphorylation of H2AX after exposure of HSPCs to AAV (4 different donors) alone or with DSB repair by AAV or ODN. n = 4 experiments. **P < .01, ****P < .0001, paired Student t test. N, naive; SSC, side scatter.
To assess phagocyte-restricted gp91phox expression, myeloid-differentiated X-CGD CD34+ HSPCs gene-edited with CYBB E7-13pA exhibited much lower gp91phox expression compared with ODN mutation–repaired cells (Figure 3B-C). The bovine growth hormone polyadenylation (pA) signal in the CYBB E7-13pA donor template was inferior compared with the endogenous CYBB 3′ untranslated region, but the expression improved with the addition of a WPRE (Figure 3B).19
Oligonucleotide donor repair of CYBB c.676 C>T in X-CGD HSPCs in vitro
Finally, we sought to optimize specific correction of CD34+ HSPCs from X-CGD patients with the c.676 C>T mutation, which accounts for ∼6% of patients with X-CGD. The addition of i53 with Cas9 mRNA doubled the efficiency of ODN-mediated gene mutation repair to achieve 64% gp91phox+ cells, compared with ∼75% gp91phox+ HD normal control (Figure 3D, top panel). Functional restoration of NOX2 activity showed a corresponding increase in DHR+ cells (Figure 3D, bottom panel), in which DHR levels for genome-edited X-CGD patient and HD cells were comparable, indicating normalized NOX2 activity after genome editing.
Genome editing with Cas9 mRNA or RNP
Because the HDR-enhancing agents were delivered as mRNA, comparisons were made by using Cas9 delivered as mRNA plus sgRNA or as RNP of Cas9 protein complexed with sgRNA, both Streptococcus pyogenes derived. Surprisingly, despite the recent report of higher toxicity related to Cas9 mRNA due to upregulation of interferon-related immune responses,20 higher gene correction efficiencies were observed with Cas9 mRNA (68% vs 47% respectively) (Figure 3E). This finding may be related to the specific design of Cas9 mRNA to avoid innate immune responses used in this study. Using Cas9 mRNA with i53, a highly efficient gene mutation repair was observed that resulted in percentages of gp91phox+ cells comparable with HD (Figure 3D) and superior to Cas9 mRNA without i53 or RNP with i53 (Figure 3F). We confirmed conversion of the mutation to normal sequence by high throughput sequencing (HTS) with 18 000 to 20 000 reads per sample, in which the percentages of corrected alleles were twofold higher with i53, at 51.7% (range, 42%-56%) for Cas9 mRNA and 38% for RNP compared with 17% without i53 (Figure 3G).
DNA damage response
To evaluate for potential cellular responses to genome editing, phosphorylated H2AX (γH2AX) was assessed as a surrogate for DNA damage response. A massive (>70% γH2AX+ cells) DDR was elicited early (2 hours) by exposure to AAV even without DNA cleavage (Figure 3H); this increased with time (Figure 3I), as observed with 4 AAV donors, indicating a class effect of AAV21,22 that was not similarly observed with ODN (Figure 3J). This finding is consistent with previous reports showing that AAV triggers the activation of ataxia-telangiectasia–mutated and Rad3-related protein (ATR) kinase, which induces the phosphorylation of H2AX.
Transplant of genome-edited CD34+ HSPCs: kinetics, long-term engraftment, and stability
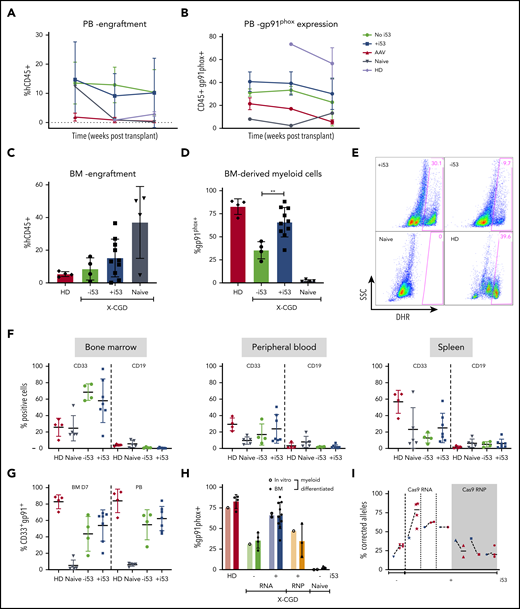
To assess the durability of the mutation-repaired HSPCs, CD34+ HSPCs from X-CGD patients with a CYBB c.676 C>T mutation underwent gene repair with ODN (±i53) or by TI with AAV CYBB E7-13pA donor (+i53); they were transplanted into 6- to 8-week-old immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice for 18 to 26 weeks. Serial peripheral blood analysis at 8, 12, and 26 weeks showed comparable engraftment rates with or without i53 (Figure 4A) but consistently higher percentages of gp91phox+ cells for ODN +i53 repaired cells (Figure 4B). Disappointingly, TI using an AAV CYBB E7-13pA donor resulted in poor engraftment, which precludes meaningful interpretation of gp91phox expression following transplantation; however, low engraftment of HD cells suggests there may be other confounding factors to impair engraftment in this study. Despite some reports of robust engraftment of CRISPR/AAV genome-edited peripheral blood CD34+ HSPCs,12,23 many studies have reported poor engraftment, particularly for diseases in which corrected cells do not have a survival advantage.24-26 A likely explanation is the massive p53 activation after exposure to AAV, as indicated by the γH2AX expression that was further exacerbated by Cas9/sg–mediated DSB, as shown earlier (Figure 3H-J). We observed robust engraftment of ODN-edited cells with human CD45+ (mean of 11.1% without i53 and 14.0% with i53) in mice bone marrow (n = 15 mice; 4 independent experiments, three X-CGD patients with CYBB c.676 C>T mutation) (Figure 4C). The results for phenotype correction were even more striking in the i53 group, in which myeloid-differentiated CD45+ cells sorted from transplanted mice bone marrow revealed unprecedented levels of gp91phox+ cells up to 82% (36%-82%; mean, 66%; n = 7 mice, 4 experiments from 3 patients) (Figure 4D). Such levels were comparable with those of healthy controls (80%-87%), and significantly higher than the group without i53 (mean, 35%). Corresponding DHR analysis confirmed restored NOX2 activity in i53-aided genome-edited cells to levels comparable to those of HDs and superior to genome editing without i53 (Figure 4E). The lineage composition (CD33+ myeloid and CD19+ B lymphoid) in the transplanted mice bone marrow, peripheral blood, and spleen were not affected by i53 (Figure 4F), and the gp91phox expression in bone marrow in vitro differentiated CD33+ myeloid cells is comparable to that in peripheral blood CD33+ myeloid cells (Figure 4G).
Highly efficient correction of engrafting X-CGD HSPCs by SpCas9/sg mRNA/ODN donor with i53. (A) Peripheral blood (PB) from mice transplanted with X-CGD HSPCs GE with ODN or AAV (CYBB E7-13pA) analyzed at weeks 8, 12, and 26 for engraftment (hCD45+). (B) Percentages of gp91phox+ cells in hCD45+ PB. (C) FACS showing human CD45+ engraftment in mice bone marrow (BM) 18 to 26 weeks after transplant with ODN mutation-repaired X-CGD HSPCs (n = 3 patients). (D) Gp91phox expression in myeloid-differentiated BM human CD45+ cells from NSG transplanted with ODN mutation–repaired X-CGD HSPCs (3 patients). (E) DHR assay showing ROS production in the myeloid-differentiated cells in panel D. (F) Lineage (CD33+ and CD19+) composition in BM, PB, and spleen of transplanted mice. (G) Gp91phox expression in CD33+ myeloid cells in myeloid-differentiated BM and PB. (H) Percentages of gp91phox+ before (in vitro) and after transplant (BM) with ODN-treated cells using Cas9 RNA (±i53) vs RNP (n = 4 experiments; 3 X-CGD patients, 11 mice). (I) Correction rates (% corrected alleles) before (input, 5 days post-electroporation, blue) vs after transplant (red) (n = 3 X-CGD patients). **P < .001.
Highly efficient correction of engrafting X-CGD HSPCs by SpCas9/sg mRNA/ODN donor with i53. (A) Peripheral blood (PB) from mice transplanted with X-CGD HSPCs GE with ODN or AAV (CYBB E7-13pA) analyzed at weeks 8, 12, and 26 for engraftment (hCD45+). (B) Percentages of gp91phox+ cells in hCD45+ PB. (C) FACS showing human CD45+ engraftment in mice bone marrow (BM) 18 to 26 weeks after transplant with ODN mutation-repaired X-CGD HSPCs (n = 3 patients). (D) Gp91phox expression in myeloid-differentiated BM human CD45+ cells from NSG transplanted with ODN mutation–repaired X-CGD HSPCs (3 patients). (E) DHR assay showing ROS production in the myeloid-differentiated cells in panel D. (F) Lineage (CD33+ and CD19+) composition in BM, PB, and spleen of transplanted mice. (G) Gp91phox expression in CD33+ myeloid cells in myeloid-differentiated BM and PB. (H) Percentages of gp91phox+ before (in vitro) and after transplant (BM) with ODN-treated cells using Cas9 RNA (±i53) vs RNP (n = 4 experiments; 3 X-CGD patients, 11 mice). (I) Correction rates (% corrected alleles) before (input, 5 days post-electroporation, blue) vs after transplant (red) (n = 3 X-CGD patients). **P < .001.
Because failure to engraft is a primary concern for genome editing of peripheral blood CD34+ HSPCs,24,25 we compared pretransplant input samples (in vitro) vs corresponding samples after transplant (‘BM’) for gp91phox expression (Figure 4H) and frequency of corrected alleles (Figure 4I). Gp91phox expression was similar in input and posttransplant samples after myeloid differentiation. Targeted HTS of the human CD45+ cells sorted from transplanted mice bone marrow revealed 45% to 75.8% (RNA) and 17% to 32% (RNP) corrected alleles. Remarkably, the levels of correction were maintained in Cas9 mRNA–repaired cells but decreased in RNP-repaired cells by 26 weeks after transplantation. Because functional NOX2 does not confer a survival advantage, a possible explanation is that the true long-term engrafting HSPCs were corrected at a higher rate than the total HSPCs when edited with Cas9 mRNA compared with RNP. Overall, the persistence of repaired alleles 18 to 26 weeks’ posttransplant shows that viable long-term correction of HSPCs is possible at engraftment levels many fold higher than clinical target correction levels of >10%.
Molecular safety analysis
We next performed long-range PCR to detect potential large deletions after editing (Figure 5A-B; supplemental Figures 1-3). Large deletions were observed following DSBs alone27 ; these increased with i53, which inhibits the dominant NHEJ DSB repair pathway. However, providing an ODN donor eliminated most of the deletions, even in the presence of i53.
![Molecular analysis of treated X-CGD HSPCs at on-target (ON) and OT sites confirm the high specificity of the SpCas9/sg/ODN genome-editing system maintained in the presence of i53. (A) Long-range PCR at CYBB E7 target site in HSPCs (naive, Cas9/sg alone, or with i53, ODN donor, or both). Genomic deletions shown by loss of coverage of sequencing reads. (B) Graph summarizing frequency of large deletions in samples treated as indicated. (C) ON and OT sites identified by circularization for high-throughput analysis of nuclease genome-wide effects by sequencing (CHANGE-seq) with mismatches as shown, with the number of reads for respective sites as indicated on the right. (D) Manhattan plot of the ON and OT sites with the sg7 sgRNA on CYBB c.676 C>T mutation X-CGD patient genomic DNA. The arrow indicates CYBB on chromosome X. (E) Indel frequency at ON and OT sites before (input) and after transplant (26 weeks) in NSG (output) of HSPCs (naive [gray]), GE with (blue) or without (red) i53 (mean ± standard deviation, ordinary one-way analysis of variance, not significant). (F) Percentages of reads with size discrepancies indicating indels for naive (N), or treated with (+) or without (-) i53 at ON and at each OT site. (G) Frequency of DSB repair events in vitro (IV) and posttransplant (fresh BM) with/without i53.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/19/10.1182_blood.2020008503/1/m_bloodbld2020008503f5.png?Expires=1768894606&Signature=n0AT63zNFb4qBqsdkqngW0Db1n7tfEJ-NSQxCMHRFAhno9EKtilpZJfqucZBJ1dQrNfWGbk61go-KgdT6FXcgdc5C5hwqb6k3wNTemB-gDr0MRz7XtBd4yF1K-5PEMFgBl0CR6LpwSMI2N9N6Z8aOvXD9O06QdTkNu-OqP5v4ISJkdRnAUFO3q1ReCpKU3QS7FTQQao6~8weacChu6SAnwftEr3AOtRZT4Q2o86XFc4MFT-W1sqLzhObKd1VrPtwnItmRrcMBzqhN4z6bk8cd-uOAyzN4VaiCIoHNKknM4R0-7yM9V3qDNyOAm2Uiw-97G-mV2HK0e7Dlsdm8eLODQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Molecular analysis of treated X-CGD HSPCs at on-target (ON) and OT sites confirm the high specificity of the SpCas9/sg/ODN genome-editing system maintained in the presence of i53. (A) Long-range PCR at CYBB E7 target site in HSPCs (naive, Cas9/sg alone, or with i53, ODN donor, or both). Genomic deletions shown by loss of coverage of sequencing reads. (B) Graph summarizing frequency of large deletions in samples treated as indicated. (C) ON and OT sites identified by circularization for high-throughput analysis of nuclease genome-wide effects by sequencing (CHANGE-seq) with mismatches as shown, with the number of reads for respective sites as indicated on the right. (D) Manhattan plot of the ON and OT sites with the sg7 sgRNA on CYBB c.676 C>T mutation X-CGD patient genomic DNA. The arrow indicates CYBB on chromosome X. (E) Indel frequency at ON and OT sites before (input) and after transplant (26 weeks) in NSG (output) of HSPCs (naive [gray]), GE with (blue) or without (red) i53 (mean ± standard deviation, ordinary one-way analysis of variance, not significant). (F) Percentages of reads with size discrepancies indicating indels for naive (N), or treated with (+) or without (-) i53 at ON and at each OT site. (G) Frequency of DSB repair events in vitro (IV) and posttransplant (fresh BM) with/without i53.
Molecular analysis of treated X-CGD HSPCs at on-target (ON) and OT sites confirm the high specificity of the SpCas9/sg/ODN genome-editing system maintained in the presence of i53. (A) Long-range PCR at CYBB E7 target site in HSPCs (naive, Cas9/sg alone, or with i53, ODN donor, or both). Genomic deletions shown by loss of coverage of sequencing reads. (B) Graph summarizing frequency of large deletions in samples treated as indicated. (C) ON and OT sites identified by circularization for high-throughput analysis of nuclease genome-wide effects by sequencing (CHANGE-seq) with mismatches as shown, with the number of reads for respective sites as indicated on the right. (D) Manhattan plot of the ON and OT sites with the sg7 sgRNA on CYBB c.676 C>T mutation X-CGD patient genomic DNA. The arrow indicates CYBB on chromosome X. (E) Indel frequency at ON and OT sites before (input) and after transplant (26 weeks) in NSG (output) of HSPCs (naive [gray]), GE with (blue) or without (red) i53 (mean ± standard deviation, ordinary one-way analysis of variance, not significant). (F) Percentages of reads with size discrepancies indicating indels for naive (N), or treated with (+) or without (-) i53 at ON and at each OT site. (G) Frequency of DSB repair events in vitro (IV) and posttransplant (fresh BM) with/without i53.
Low OT rates not affected by i53-mediated HDR enhancement
For a comprehensive screen for potential OT sites in a genome-unbiased manner, we performed circularization for high-throughput analysis of nuclease genome-wide effects by sequencing (CHANGE-seq) on genomic DNA from human CD34+ cells.27 A highly specific profile was revealed (specificity ratio, 0.75), with 32 potential OT candidates identified (Figure 5C-D), all in noncoding regions. Reassuringly, HTS of the target site and the top 6 most frequent OT sites confirmed very low (<1%) indels at the OT sites in the pretransplant genome-edited HSPCs (input) and after transplant (output; human CD45+ from the mouse bone marrow) (Figure 5E) that were no different from untreated cells. In addition, comparison of pretransplant and posttransplant samples further confirmed the maintenance of i53-enhanced correction rates (∼75%) with i53 (Figure 4I). By comparing amplification product sizes that are more sensitive, we also did not detect significant number of indels at the 6 OTs (Figure 5F).11
Finally, preclinical toxicity studies monitoring hematology, chemistry, and weights of transplanted mice were also unremarkable (supplemental Figure 3). To detect potential re-cutting of a repaired DSB, insertions of one or more bases from a first cut should result in one or more bases before or after the mutation “A.” In contrast, a re-cutting event of a repaired mutation (from T/A to G/C) should result in an insertion before or after a “G” (Figure 5G). Very low re-cutting events causing insertions, especially from bone marrow of <0.1% with use of i53, were observed, suggesting that the rates of re-cuts were at least 1000-fold less than HDR. To further address potential concerns regarding genomic instability following transient inhibition of 53BP1, we performed SKY karyotyping and FISH studies on cells GE with (10 cells) or without (15 cells) i53 (Figure 6). This limited characterization showed no evidence of translocations or chromosomal errors.
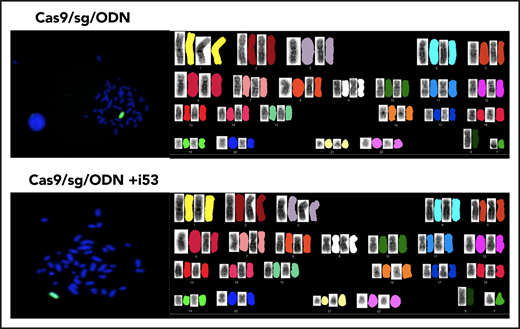
FISH (left) and karyotyping (right) studies in GE HSPCs. HSPCs CD34+ were genome edited without i53 (top) (n = 10 cells) and with i53 (bottom) (n = 15 cells).
FISH (left) and karyotyping (right) studies in GE HSPCs. HSPCs CD34+ were genome edited without i53 (top) (n = 10 cells) and with i53 (bottom) (n = 15 cells).
Discussion
The current study sought to optimize functional correction levels in long-term engrafting CD34+ HSPCs necessary for clinical translation of HDR-mediated targeted gene correction for X-CGD. X-CGD carriers with low percentages of normal NOX2 protein–producing (DHR+) cells are asymptomatic compared with X-CGD patients with missense mutations capable of producing low amounts of NOX2 protein. An important implication for X-CGD gene therapy reported here is the critical importance of achieving near-normal levels of NOX2 per cell in providing a protective effect. The recent ex vivo lentivector gene therapy for X-CGD inserts one or more copies of a codon-optimized full-length CYBB cDNA into CD34+ HSPCs,4 with the expression driven by an engineered chimeric promoter that is phagocyte specific28 but unfortunately attains only ∼33% of per-cell levels of DHR by mean fluorescent intensity and cytochrome c reduction levels compared with healthy donors.4 To retain physiological regulation of CYBB expression by the CYBB promoter, we assessed functional gene correction strategies of HDR-mediated gene repair of the c.676 C>T mutation using an ODN donor template or targeted insertion of exon 7-13 cDNA in the CYBB locus.
Functional gene correction strategies require careful consideration of the cut/repair site and the donor design, particularly for TI. We observed two main issues with our exon 7-13 cDNA TI strategy in this report. The first is the relatively low protein production despite capturing the endogenous promoter for regulation, likely attributable to other important mechanisms such as posttranscriptional regulation at the 3′ terminus. The addition of regulatory elements such as WPRE may aid in resolving this issue.19 A second issue that has plagued the targeted genome editing field in general is the profound loss of corrected cells in long-term engrafted CD34+ HSPCs, particularly for TI using AAV donor templates,24-26 which was observed in this report as well. Our data with H2AX expression exhibited a profound impact of AAV on HSPCs that is not observed with ODN donors. However, there is considerable progress in the field toward controlling DNA damage responses to genome editing,11,26 which may significantly improve this issue. However, in the interim, our current data for gene repair of the c.676 C>T mutation with an ODN donor template show highly efficient long-term correction at levels comparable to those of healthy controls, and it is now poised for clinical application.
Excellent characterization of DNA repair pathways has identified multiple HDR-enhancing agents. Our data show significant increases in gene correction rates achieved through transient suppression of 53BP1 activity. After DSB, competing repair pathways (NHEJ and HDR) are regulated by the epigenetic reader, 53BP1, which binds to demethylated lysine 20 on histone 4 that is recruited to DSBs. The binding occurs at the TTD of 53BP1, and interference at this critical site can be achieved by agents such as i53 (an ubiquitin variant) that impairs recruitment of 53BP1 to DSBs and NHEJ. In this report, we used i53 mRNA, which can be added to the genome-editing reagent cocktail for electroporation into cells.
Strategies for enhancing DNA repair during GE give rise to concerns of potentially increasing mutagenesis. The OT indels in pretransplant and long-term engrafting cells after 18 to 26 weeks’ transplant were similarly low at <1% and not statistically different from untreated samples, providing reassurance that Cas9 mRNA or i53 did not increase OT mutations. The editing efficiencies with Cas9 mRNA were higher than those of RNP, despite an earlier report of increased interferon-related immune responses associated with Cas9.20 This finding could be attributable to the difference in mRNA preparations specially designed to avoid innate immune responses. Despite concerns for specificity with Cas9 mRNA with increased OT mutations, we observed no significant OT indels when edited in the absence or presence of i53 above baseline in nontreated cells, suggesting that the prevailing determining factor to editing specificity may be intrinsic to the guide RNA used. The high specific nature of this CYBB c676 Sg7 accounts for the extremely low re-cutting rates of repaired alleles and absence of cutting activity when Sg7 was applied to healthy volunteer cells with a single bp difference at the mutation site.
NHEJ inhibition using i53 potentially affects genomic stability. Long-range PCR identified large (>1000 bp) deletions (0.26%) following DSB repair in the absence of a repair template, as previously reported29 (Figure 5A; supplemental Figure 1). Providing a donor facilitates HDR and significantly reduces the frequency of large deletions to a rare occurrence (0.01%). Inhibition of 53BP1 increased the rate of large deletions (7.08%) but, in the presence of a repair template, remains rare at 0.01% (without i53) to 0.04% (with i53). Overall, the frequencies of HDR at target site are at the ∼50% range, >1000-fold higher than the large deletions at <0.01%. Additional studies are necessary to better assess the impact of i53 in this gene-editing approach as well. Results of our limited normal karyotyping and FISH studies are encouraging, but more sensitive methods are needed to detect infrequent genotoxic events. No tumors have been detected in our mice transplant studies to date. These potential risks associated with use of i53 should be balanced with the benefits of a highly efficient gene correction rate in long-term engrafting HSPCs, compared with the alternate lentivector gene therapy option that results in completely random insertion of transgenes that lack endogenous regulation for physiological expression.
The biggest barrier to translating HDR-dependent functional gene correction strategies to clinical treatment of patients lies in the challenges in attaining stable correction in long-term engrafted HSPCs. Our data show that our optimized protocols with i53 enhancement of HDR for ODN-based mutation repair resulted in efficient gene editing of peripheral blood CD34+ HSPCs that was maintained at 18 to 26 weeks’ transplant, indicating stable correction of long-term engrafting HSPCs to achieve gp91phox expression at rates comparable to those of HD controls. Further studies would be helpful to verify increased editing of long-term HSPC with i53. This highly efficient nonviral approach to mutation repair is comparatively inexpensive, relatively easy to perform, and well poised for clinical translation for the patients who share this specific mutation. However, additional scale-up studies are needed to confirm that these outcomes are translatable at larger scales for clinical application. If so, this versatile approach would also be applicable for repair of other genetic diseases caused by single or few mutations, such as sickle cell disease.25
All necessary information for reagents is provided in the article. The MaxCyte electroporation device is used under a licensing agreement. Requests for original data should be submitted to Suk See De Ravin (sderavin@niaid.nih.gov).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and healthy donors for their contribution to this study and the Department of Transfusion Medicine at the National Institutes of Health Clinical Center for their collection and processing of CD34+ HSPCs.
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health, under intramural project numbers Z01-Al-00644 and Z01-Al-00988.
Authorship
Contribution: S.S.D.R. conceived and wrote the manuscript; C.L.S. and J.B. edited the manuscript; S.S.D.R., L.L., J.B., T.L., and N.T. performed experiments in human CD34+ HSPCs and transplant studies, and analyzed data; S.L., X.W., C.R.L., G.L., S.T., and B.P.K. performed sequencing and OT analyses; U.C. and S.M.K. assisted with animal studies; D.B.K. assisted with NADPH oxidase assays and analysis; M.P.-D., L.L., and S.S.D.R. designed donor templates; S.S.B. performed karyotyping and FISH studies; R.J.M., A.B.C., and G.A.D. produced mRNAs; and S.H., M.H.P., and H.L.M. provided guidance.
Conflict-of-interest disclosure: L.L. was a full-time employee of MaxCyte Biosystems at the time of this study. R.J.M., A.B.C., and G.A.D. are full-time employees of CELLSCRIPT, LLC. B.P.K. is an inventor on patent applications filed by Mass General Brigham that describe genome engineering technologies. The remaining authors declare no competing financial interests.
Correspondence: Suk See De Ravin, CRC, 5W-3817, Center Dr, Bethesda, MD 20892; e-mail: sderavin@niaid.nih.gov; and Harry L. Malech, CRC, 5W-3750, Center Dr, Bethesda, MD 20892; e-mail: hmalech@niaid.nih.gov.