TO THE EDITOR:
Previously untreated follicular lymphoma (FL) is an indolent disease with a median survival of more than 18 years.1 However, a subset of patients with FL who relapse early has significantly worse outcomes.2 Identification of such high-risk patients is critical to help predict which patients will benefit most from novel therapies.
The follicular lymphoma international prognostic index (FLIPI) is an early classification metric to identify a high-risk subset of FL.3 Additional clinical-based models, such as FLIPI-24,5 and the PRIMA trial prognostic index (PRIMA-PI)6 have shown limited improvement over FLIPI. Other high-risk scores have focused on combining clinical factors with genomic biomarkers, such as the genetic mutations in the m7-FLIPI model.7 In addition, a variety of gene expression–based metrics have been published.8-10 However, all published prognostic models were developed and validated in patients treated with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP)– or cyclophosphamide, vincristine, and prednisone (CVP)–like regimens, with or without rituximab (R). Recent studies have suggested that these models reflect biological differences in response to standard chemotherapy regimens5,11,12 and that their utility may not extend to other therapies such as obinutuzumab (G) or bendamustine.
We evaluated the performance and treatment dependence of the published gene expression–based models in patients with first-line FL treated in the randomized, phase 3 GALLIUM study with G-chemo or R-chemo (CHOP, CVP, or bendamustine) (#NCT01332968).
The GALLIUM study design and patient population have been previously described.13 The primary study end point was investigator-assessed progression-free survival (PFS). GALLIUM was conducted in accordance with the International Conference on Harmonization guidelines for Good Clinical Practice. The protocol was approved by the ethics committees of participating centers. All patients provided written informed consent, and genetic testing was performed on patients who provided consent for genetic analysis of their previously stored biopsy samples.
Purified RNA was used to create cDNA libraries that were assayed using TruSeq (Illumina) RNAseq to measure gene expression (supplemental Methods, available on the Blood Web site), and we examined a total of 5 previously published signatures: the PRIMA 23-gene signature and the ICA13 signature from Huet et al10 ; the 6-gene T-effector signature (CD8A, EOMES, GZMA, GZMB, IFNγ, PRF1), described originally in Bolen et al9 ; and the IR1 and IR2 signatures from Dave et al.8
To quantify the expression of the PRIMA 23-gene signature, published Nanostring gene weights were used10 to generate a weighted sum of the 23 genes, and high risk was defined as the top 25th percentile. For the other signatures, we used a principal component analysis–based approach to calculate a summary score, and a high-risk definition was evaluated at 3 different cutoff points based on quartiles (25th, 50th, 75th percentile). Association with PFS was performed using a Cox proportional hazards model with FLIPI, sex, and geographic region included as covariates.
The intent-to-treat (ITT) FL population comprised 1202 patients. RNA samples were available for 372 FL patients; 98 samples were excluded because of errors during the RNA library preparation (supplemental Figure 1). The 274 patients in the final biomarker-evaluable (BE) population were consistent with the overall ITT population for clinical characteristics and outcomes, with the exception of race and choice of chemotherapy (higher prevalence of bendamustine; supplemental Table 1). Among the ITT population, use of bendamustine was significantly more common within North America and Australia, in older patients, patients with a Charlson comorbidity index ≥1, and in patients without bulky disease; consistent trends were observed among the BE population (supplemental Table 2).
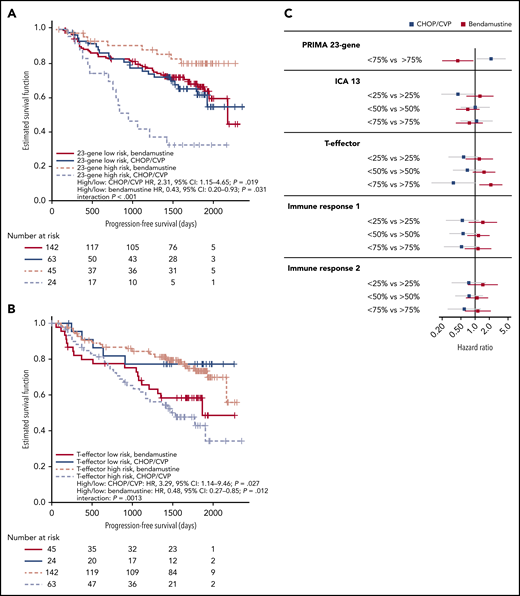
Among the 274 patients analyzed by RNASeq, we observed no prognostic effect for any of the gene signatures in the overall dataset or when patients were split by antibody treatment arm (R vs G; supplemental Figure 2). However, when split by chemotherapy regimen, a significant interaction was observed between PFS and biomarker-high status, as defined by the PRIMA 23-gene signature (interaction, P < .001; Figure 1A) and for the bottom 75th percentile of the T-effector signature (interaction P = .0013; Figure 1B). CHOP- or CVP-treated patients displayed a significantly higher risk of disease progression or death for both the 23-gene signature (P = .019) and T-effector signature (P = .027) biomarker high groups, whereas bendamustine-treated patients were at significantly lower risk (23-gene, P = .031; T-effector, P = .01). Patients classified at low risk of treatment failure by the 23-gene signature appeared to benefit equally from CHOP/CVP or bendamustine (Figure 1C).

High-risk gene signatures are differentially prognostic for PFS in patients treated with bendamustine vs CHOP/CVP. Summary scores were calculated for 5 published gene signatures. High risk for (A) the PRIMA 23-gene signature was defined as patients in the top 25th percentile or (B) the top 75th percentile for the T-effector signature. Patients were split by high-risk status and chemotherapy group, and PFS was plotted as a Kaplan-Meier curve. HR and P value for an interaction term are included. (C) High-risk definitions for gene signatures were evaluated at 3 different quartile cutoffs (25th, 50th, and 75th percentiles). PFS HRs and 95% CIs in the CHOP/CVP cohort (blue) or the bendamustine cohort (red) are plotted. CI, confidence interval; HR, hazard ratio.
High-risk gene signatures are differentially prognostic for PFS in patients treated with bendamustine vs CHOP/CVP. Summary scores were calculated for 5 published gene signatures. High risk for (A) the PRIMA 23-gene signature was defined as patients in the top 25th percentile or (B) the top 75th percentile for the T-effector signature. Patients were split by high-risk status and chemotherapy group, and PFS was plotted as a Kaplan-Meier curve. HR and P value for an interaction term are included. (C) High-risk definitions for gene signatures were evaluated at 3 different quartile cutoffs (25th, 50th, and 75th percentiles). PFS HRs and 95% CIs in the CHOP/CVP cohort (blue) or the bendamustine cohort (red) are plotted. CI, confidence interval; HR, hazard ratio.
When considered individually, a significant chemotherapy-dependent interaction was observed for 7 of the 23-gene signature genes: ABCB1, FOXO1, SEMA4B, AFF3, PRDM15, ALDH2, and KIAA0040. All genes trended toward a differential prognostic signal, and no genes consistently identified a high-risk subset across all chemotherapy regimens (Figure 2).

Evidence of differential prognostic association across all subsets of the 23-gene signature. The association of each gene was tested among patients treated with either CHOP/CVP (blue) or bendamustine (red). PFS HRs and 95% CIs from a multivariate Cox proportional-hazards model are plotted. *Demonstrated significant chemotherapy dependence (interaction, P < .05). CI, confidence interval; HR, hazard ratio.
Evidence of differential prognostic association across all subsets of the 23-gene signature. The association of each gene was tested among patients treated with either CHOP/CVP (blue) or bendamustine (red). PFS HRs and 95% CIs from a multivariate Cox proportional-hazards model are plotted. *Demonstrated significant chemotherapy dependence (interaction, P < .05). CI, confidence interval; HR, hazard ratio.
This study demonstrated that choice of chemotherapy has a significant impact on the prognostic significance of previously published gene expression signatures, with significantly worse PFS for patients defined as high risk receiving CHOP/CVP compared with those receiving bendamustine. Results were consistent across the tested genes.
The chemotherapy dependence of the high-risk signatures is surprising but not without precedent. The IR2 signature, which contains a number of genes related to macrophage infiltration, was initially predictive of worse outcomes; however, in later trials with uniform R-CHOP therapy, the IR2 signature and tumor-associated macrophages were found to be positively prognostic.9,14 Macrophage infiltration analysis in patients treated with either R-CHOP or R-CVP suggested that this was because of the addition of doxorubicin, which may be dependent on the presence of activated macrophages for efficacy.15 The weak association observed of the IR2 signature with chemotherapy is in line with these previous observations, although it is not as striking as previously seen.
Although some of the genes in the 23-gene signature are likely to be associated with macrophages, they cover a wide range of different biologic pathways. Similarly, the 6-gene T-effector signature and the IR1 signature both represent other aspects of the immune microenvironment, suggesting that these are not specifically associated with the activity of doxorubicin. The contribution of the immune microenvironment to chemotherapy response has not been well studied. Given the number of different chemotherapy drugs that vary across these patients, it is possible that each of these drugs may be dependent on a specific pathway or infiltrating immune subset for activity and that each differential prognostic effect reflects the addition or loss of one (or more) of these drugs.
One potential confounding factor is the lack of randomization of chemotherapy. Although the study was designed to limit confounding effects by asking centers to choose their chemotherapy regimen upfront, we observed a number of imbalances related to chemotherapy choice, including age, geographic region, and bulky disease status. Individually, none of these are associated with the expression of our gene signatures, and in multivariate models they do not affect the observation of a treatment effect in these patients. However, it is still possible that there are additional underlying genetic or clinical factors, such as m7FLIPI status or disease stage,16 that may affect the prognostic value of these biomarkers. To address these possible confounders, it will be necessary to validate these observations in additional datasets or clinical trials evaluating novel therapies.
These results provide new insights in interpreting prognostic gene signatures in FL and highlight the challenges of building high-risk signatures for patients, independent of treatment.
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on Roche's criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and their families, study investigators, coordinators, and nurses for taking part in GALLIUM.
Editorial support, under the direction of the lead author, was provided by Louise Profit of Ashfield MedComms, an Ashfield Health company and was funded by F. Hoffmann-La Roche Ltd. GALLIUM was sponsored by F. Hoffmann-La Roche Ltd.
Authorship
Contribution: C.R.B. wrote the first draft of the manuscript; C.R.B., G.S., J.M.V., M.Z.O., and R.M. conceived and designed the study; G.S., M.H., M.Z.O., and W.K. collected and assembled the data; C.R.B., F. Mattiello, F. Mir, J.M.V., M.Z.O., O.W., S.H., T.N., and W.H. provided data analysis and interpretation; and all authors critically reviewed the manuscript for scientific content and approved the final version for submission.
Conflict-of-interest disclosure: C.R.B. is an employee at Genentech, Inc. and has equity ownership in Roche. F. Mattiello is an employee of Roche. M.H. reports research funding from Roche and consultancy fees from Celgene, Gilead, and Roche. W.H. reports honoraria fees from Roche, Amgen, Gilead, Janssen, and Celgene; research funding from Roche, Amgen, Janssen, and Bayer; and consulting, speaker bureau, and travel fees from Roche, Amgen, Janssen, and Gilead. W.K. reports honoraria from Roche and Amgen; consulting fees from Takeda, Celgene, and Roche; and research funding from Roche, Amgen, Takeda, and Regeneron Pharmaceuticals. R.M. reports consulting fees and honoraria fees from Gilead and speaker bureau, honoraria, and travel fees from Roche/Genentech, Inc. G.S. consults or advises for AbbVie, Autolus, Celgene, Epizyme, Roche, Gilead Sciences, Janssen Pharmaceuticals, Karyopharm, Kite, Merck, MorphoSys, Novartis, Servier, and Takeda; has received honoraria from AbbVie, Amgen, BMS, Celgene, Epizyme, Roche, Gilead Sciences, Janssen Pharmaceuticals, Karyopharm, Kite, Merck, MorphoSys, Novartis, Servier, and Takeda; is a member of advisory boards for AbbVie, Autolus, Celgene, Roche, Gilead Sciences, Janssen Pharmaceuticals, Karyopharm, Kite, Merck, MorphoSys, Novartis, Servier, and Takeda; and participates in educational events for AbbVie, Amgen, Celgene, Roche, Gilead Sciences, Janssen Pharmaceuticals, Karyopharm, Kite, Merck, MorphoSys, Novartis, Servier, and Takeda. F. Mir is a former employee at Roche. O.W. reports consulting fees from Roche and Epizyme and research funding from Roche and Novartis. T.N. is an employee and has stock ownership in Roche. M.Z.O. is a former employee at Roche with patent royalties in Roche and is an employee of Novo Nordisk. J.M.V. is an employee of Foundation Medicine Inc. and former employee of Roche/Genentech, Inc. S.H. declares no competing financial interests.
Correspondence: Christopher R. Bolen, Bioinformatics & Computational Biology, Genentech Inc., One DNA Way, MS 444A, South San Francisco, CA 94080; e-mail: bolen.christopher@gene.com.