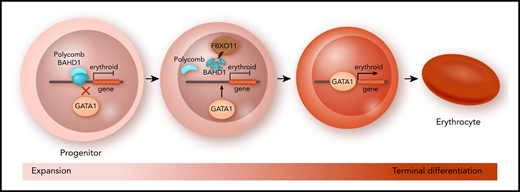
In this issue of Blood, report on a novel regulator of red cell differentiation, F-box only protein 11 (FBXO11), that removes transcriptional repressors from erythroid genes releasing their expression by targeting the chromatin-associated protein bromo adjacent homology domain–containing 1 (BAHD1) for degradation.1
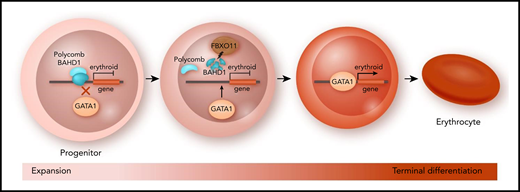
FBXO11-mediated protein degradation enables red cell differentiation. In expanding red cell progenitors, differentiation-associated genes are repressed by epigenetic factors such as the polycomb complex. To enter terminal differentiation, the E3 ubiquitin ligase FBXO11 initiates the proteasomal degradation of chromatin factor BAHD1. As a result, repressive factors are no longer recruited to erythroid genes, which allows GATA1 to activate red cell genes required for terminal differentiation.
FBXO11-mediated protein degradation enables red cell differentiation. In expanding red cell progenitors, differentiation-associated genes are repressed by epigenetic factors such as the polycomb complex. To enter terminal differentiation, the E3 ubiquitin ligase FBXO11 initiates the proteasomal degradation of chromatin factor BAHD1. As a result, repressive factors are no longer recruited to erythroid genes, which allows GATA1 to activate red cell genes required for terminal differentiation.
The ubiquitin-proteasome system is the primary pathway for cellular protein degradation. E3 ubiquitin ligases bind to specific substrate proteins to add ubiquitin groups. These posttranslational modifications signal the proteasome to degrade the substrate proteins. Hematopoietic progenitor cells differentiating toward highly specialized red cells undergo dramatic changes in their protein content. Because red cell function relies predominantly on hemoglobin, most other proteins become unnecessary. Previous studies identified several E3 ubiquitin ligases that target these superfluous proteins for degradation. However, during erythropoiesis, progenitor cells express a large number of other E3 ubiquitin ligases with unknown targets and unknown importance for erythroid differentiation. A better understanding of the role of these E3 ubiquitin ligases in erythropoiesis would have clinical implications, for example, by accelerating the development of in vitro cultured red cells for donor-independent transfusion products. In addition, the proteasomal machinery can be hijacked to target selected proteins for degradation, thereby providing a powerful tool for research and drug development.2
Xu et al have performed a CRISPR screen that inactivates the components of the protein degradation system. They observe that FBXO11 is required for terminal erythroid differentiation, but not for proliferation of progenitor cells. FBXO11 is a subunit of the SKP1-CULLIN1-F-box E3 ubiquitin ligase complex. As a specific substrate receptor, it selects proteins destined for proteasome-mediated degradation. The million-dollar question is which proteins are targeted by FBXO11. In cells that lack FBXO11, the abundance of its target proteins should increase because of disabled FBXO11-dependent degradation. The authors investigated this by comparing messenger RNA (mRNA) and protein expression levels in control and FBXO11 knockout (KO) erythroid cells. This survey yielded several proteins, including BAHD1, with increased abundance in FBXO11 KO erythroid cells without accompanying changes in mRNA levels. Further experiments demonstrated that depletion of BAHD1 partially rescues the erythroid differentiation phenotype of FBXO11 KO cells and illustrated that BAHD1 is a key target of FBXO11. These results raise the question of how BAHD1 prevents terminal erythroid differentiation.
BAHD1 is a known chromatin-binding nuclear factor that recruits transcriptional corepressors.3 Xu et al report that histone acetylation is reduced at the promoters of genes with decreased expression in FBXO11 KO cells. This result is consistent with increased BAHD1-mediated recruitment of histone deacetylases at these downregulated genes. In addition, BAHD1 depletion in FBXO11 KO cells restores the expression of many erythroid genes that are controlled by the GATA1 transcription factor, a key regulator of erythroid differentiation.4 By using chromatin immunoprecipitation sequencing, the authors found that, in many cases, BAHD1 and GATA1 have overlapping binding sites at erythroid genes. These results led to a model in which BAHD1 interferes with the binding of GATA1 to its target genes, thus preventing their activation and blocking terminal erythroid differentiation (see figure).
How does BAHD1 find its target genes? Unlike GATA1, BAHD1 does not contain a DNA-binding domain, so it cannot recognize its target genes via specific DNA elements. Instead, it binds to a repressive chromatin modification, namely triple methylation of the twenty-seventh lysine residue of histone subunit H3 (H3K27me3).5 This epigenetic mark is deposited by the polycomb repressor complex.6 To investigate the interplay of FBXO11-BADH1 and epigenetic modifiers, the investigators performed a second CRISPR screen in FBXO11 KO cells, this time targeting genes that encode epigenetic modifiers. This screen revealed that inactivation of several genes of the polycomb repressor system partially rescues the erythroid differentiation defect in a manner similar to that seen for BAHD1 inactivation. Furthermore, the authors demonstrated protein-protein interactions between the polycomb factor EZH2 and BAHD1. Collectively, their work provides significant new insight into how the transition from progenitors to terminal erythroid differentiation is regulated (see figure).
As with any original research, new questions arise. Because transcription levels of some genes in FBXO11 KO cells are not fully restored upon BAHD1 suppression, FBXO11 likely targets additional regulators for degradation. Identification and functional analysis of these factors would further increase our understanding of the molecular control of erythropoiesis. Follow-up studies might identify the interplay of FBXO11 and repressive complexes, other than polycomb, that facilitate the transition from progenitor cells to terminally differentiated red cells. Another question raised by the Xu et al study is how FBXO11 is activated, especially because the decline in BAHD1 protein during erythroid maturation does not coincide with rising FBXO11 levels. Future studies are warranted to test the authors’ hypothesis that a posttranslational modification of BAHD1 (eg, phosphorylation7 ) enables FBXO11-mediated ubiquitination. The role of BAHD1 in DNA methylation8 may also be part of BAHD1-mediated repression of erythroid genes; global demethylation of DNA occurs during terminal erythroid differentiation.9
Some of the other BAHD1-interacting proteins that are not studied in the article by Xu et al provide additional avenues for further investigation. For instance, the histone methyl transferases EHMT1 and EHMT2 repress fetal hemoglobin in adult red cells.10 Repurposing the FBXO11-BAHD1 axis to modulate EHMT1 and EHMT2 activity may reactivate fetal hemoglobin expression, thus providing a strategy to ameliorate the symptoms of patients with β-hemoglobinopathies. Finally, if the FBXO11-BAHD1 axis is indeed a toggle to regulate erythroid output in the bone marrow, this may have implications for the clinical course and possibly also the treatment of patients with erythroid differentiation defects such as those associated with myelodysplastic syndromes and polycythemia vera. Recent studies demonstrating the feasibility of targeted protein degradation hold promise for future drug development based on tinkering with the ubiquitin-proteasome system.2
Conflict-of-interest disclosure: The authors declare no competing financial interests.