Key Points
Platelets harbor a pool of plasminogen that is released and retained on the activated membrane via the novel platelet receptor Plg-RKT.
Platelet-derived plasminogen drives surface-mediated plasminogen activation and local fibrinolysis.

Abstract
Plasminogen activation rates are enhanced by cell surface binding. We previously demonstrated that exogenous plasminogen binds to phosphatidylserine-exposing and spread platelets. Platelets contain plasminogen in their α-granules, but secretion of plasminogen from platelets has not been studied. Recently, a novel transmembrane lysine-dependent plasminogen receptor, Plg-RKT, has been described on macrophages. Here, we analyzed the pool of plasminogen in platelets and examined whether platelets express Plg-RKT. Plasminogen content of the supernatant of resting and collagen/thrombin-stimulated platelets was similar. Pretreatment with the lysine analog, ε-aminocaproic acid, significantly increased platelet-derived plasminogen (0.33 vs 0.08 nmol/108 platelets) in the stimulated supernatant, indicating a lysine-dependent mechanism of membrane retention. Lysine-dependent, platelet-derived plasminogen retention on thrombin and convulxin activated human platelets was confirmed by flow cytometry. Platelets initiated fibrinolytic activity in fluorescently labeled plasminogen-deficient clots and in turbidimetric clot lysis assays. A 17-kDa band, consistent with Plg-RKT, was detected in the platelet membrane fraction by western blotting. Confocal microscopy of stimulated platelets revealed Plg-RKT colocalized with platelet-derived plasminogen on the activated platelet membrane. Plasminogen exposure was significantly attenuated in thrombin- and convulxin-stimulated platelets from Plg-RKT−/− mice compared with Plg-RKT+/+ littermates. Membrane exposure of Plg-RKT was not dependent on plasminogen, as similar levels of the receptor were detected in plasminogen−/− platelets. These data highlight Plg-RKT as a novel plasminogen receptor in human and murine platelets. We show for the first time that platelet-derived plasminogen is retained on the activated platelet membrane and drives local fibrinolysis by enhancing cell surface–mediated plasminogen activation.
Introduction
Platelets are a reservoir for a diverse range of proteins, including many that direct the hemostatic response. In addition, platelets are a focal point of fibrin formation because of their ability to facilitate thrombin generation when activated. Classically platelets have been described as antifibrinolytic, because of the high concentrations of PAI-1 within their α-granules,1 which is the major pool of circulating PAI-1. We have shown recently that functionally active PAI-1 is retained on the activated platelet membrane.2 Our work also describes the release of platelet-derived factor XIII-A (FXIII-A) by activated platelets, which is retained on the stimulated membrane and is functional in cross-linking of plasma-derived α2-antiplasmin (α2AP) to fibrin.3 Furthermore, platelets drive the process of clot retraction through fibrinogen binding to the integrin αIIbβ3.4-6 Retraction of clots condenses the crosslinked α2AP7 and attenuates binding of tissue plasminogen activator (tPA)8 to platelet-associated fibrin, making them more resistant to lysis than uncompacted clots.9,10
The role of platelets in regulation of fibrinolysis is multifaceted, because activated platelets also provide binding sites for plasma-derived plasminogen.11,12 We have demonstrated that plasma-derived plasminogen binds to distinct locations in different subpopulations of platelets via both fibrin-dependent and fibrin-independent mechanisms.12 Procoagulant platelets express phosphatidlyserine (PS)13 and are characterized by a balloon-type structure. They bind coagulation factors via Gla domains to promote local thrombin generation and downstream fibrin formation. Exogenous plasminogen was localized to the platelet cap12 of PS-exposing platelets. This protruding cap,14 also referred to as platelet body,15,16 is also rich in fibrinogen, thrombospondin,14 FXIII-A,3 PAI-1,2,12 and factors IXa, Xa/X, Va, and VIII.17 Adherent spread platelets expose activated αIIbβ3 and bind fibrin and other platelets preventing premature thrombus degradation.18 In this subpopulation, binding of plasma plasminogen is concentrated centrally over the granulomere.12
Under physiologic flow conditions, plasma-derived plasminogen is incorporated into the growing thrombus by binding both directly to the platelet surface and indirectly via platelet-associated fibrinogen, thus facilitating fibrinolysis.12 Our laboratory has demonstrated that the platelet surface promotes reciprocal activation of single chain urokinase (scuPA) and plasminogen via a membrane-dependent process.19
Plasminogen activation by tPA is significantly augmented by colocalization of the reactants on fibrin or cellular surfaces20,21 including platelets.12,22 Binding of plasminogen to fibrin or cells occurs via lysine binding sites in the kringle domains. Furthermore, binding to the cell surface protects plasmin from inhibition by α2AP.23-25 Several plasminogen binding proteins have been described on different cells types.26 A common feature of these plasminogen receptors is their exposure of C-terminal lysines, which promotes plasminogen binding and activation.27 Recently a novel transmembrane plasminogen receptor has been described on the surface of macrophages, which is the only known plasminogen receptor to be synthesized with a C-terminal lysine.28 This receptor has been designated Plg-RKT and has an active role in macrophage migration29 and recruitment.30-32
Platelets have been suggested to harbor plasminogen within their α-granules33,34 ; however, little is known about this pool. Here, we demonstrate for the first time that a pool of platelet-derived plasminogen is exposed and retained on the surface of activated platelets. Once stimulated, platelets promote plasminogen activation on their surface and can drive fibrinolysis. We also demonstrate the presence of the novel transmembrane receptor, Plg-RKT, on the platelet membrane, which functions to retain platelet-derived plasminogen.
Methods
Study approval
All animal experiments were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute. Experiment using human blood samples were approved by the University of Aberdeen College Ethics Review Board.
Collection of human blood and preparation of platelets
Blood was drawn from healthy volunteers according to the Declaration of Helsinki. Peripheral blood was collected in acid citrate dextrose solution A vacuettes (Greiner Bio-One Ltd, Stonehouse, United Kingdom). Platelets were isolated by centrifugation at 260g for 15 minutes to collect platelet-rich plasma (PRP). PRP was centrifuged at 870g for 15 minutes and then washed by centrifugation at 870g for 15 minutes in N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) wash buffer (10 mM HEPES, pH 6.6, 136 mM NaCl, 2.7 mM KCl, 2 mM MgCl2, 0.1% glucose, and 0.1% bovine serum albumin [BSA]) containing 0.1 U/mL apyrase (Sigma-Aldrich, Gillingham, United Kingdom) and acid citrate dextrose (80 mM trisodium citrate, 52 mM citric acid, and 183 mM glucose). Pelleted platelets were resuspended in HEPES buffer (10 mM HEPES, pH 7.45, 136 mM NaCl, 2.7 mM KCl, 2 mM MgCl2, 0.1% glucose, and 0.1% BSA) containing 0.1 U/mL apyrase. Platelet counts were performed on a Siemens ADVIA 2120i Hematology System (Camberely, United Kingdom) by the Haematology Department, Aberdeen Royal Infirmary or on Sysmex XP-300 Hematology Analyzer (Norderstedt, Germany).
Preparation of mouse platelets
Mice deficient in Plg-RKT (Plg-RKT−/−) were generated as previously described.30 Plg-RKT+/− mice were crossed to obtain Plg-RKT−/− mice and Plg-RKT+/+ littermate controls. Plasminogen-deficient (Plg−/−) mice were a kind gift from Victoria Ploplis (University of Notre Dame, South Bend, IN). All mice were in the C57Bl/6J background. Mice were anesthetized with CO2 before performing cardiac puncture. Blood was collected into 3.8% trisodium citrate. Blood was pooled from 3 age- and sex-matched mice, and the final volume was adjusted to 7 mL using modified Tyrode’s buffer (10 mM HEPES, pH 6.5, 135 mM NaCl, 2.9 mM KCl, 0.42 mM NaH2PO4, 5.5 mM glucose, 11.9 mM NaHCO3). PRP was obtained by centrifugation at 230g for 7 minutes at ambient temperature (IEC PR-7000 Centrifuge; International Equipment Co, Needham Heights, MA). PRP was centrifuged at 500g for 13 minutes in modified Tyrode’s buffer (pH 6.5) to obtain platelet pellets. The pellet was resuspended in modified Tyrode’s buffer (10 mM HEPES, pH 7.4, 135 mM NaCl, 2.9 mM KCl, 0.42 mM NaH2PO4, 5.5 mM glucose, 11.9 mM NaHCO3) containing 0.1 U/mL apyrase. Platelets counts were obtained on an IDEXX ProCytoDx Hematology analyzer.
Detection of plasminogen by enzyme-linked immunosorbent assay
Platelets (5 × 108 platelets/mL) were isolated in either presence or absence of ε-aminocaproic acid (εACA) (200 mM) and stimulated with collagen (20 µg/mL) and thrombin (100 nM) for 30 minutes. Plasminogen was detected in the supernatant using an enzyme-linked immunosorbent assay (ELISA), as described previously,35 with the exception that the detection antibody was a goat–anti-human plasminogen polyclonal antibody (0.1 μg/mL, Enzyme Research Laboratories).
Flow cytometry
Washed platelets (2 × 108 platelets/mL) in HEPES buffer, pH 7.45, (human) or modified Tyrodes buffer (mouse) were either unstimulated or stimulated in the presence of 2 mM CaCl2 with 100 ng/mL convulxin (CVX; Enzo Life Sciences) ± 20 μM thrombin receptor activator peptide-6 (TRAP-6) or 100 nM thrombin (Sigma-Aldrich) in the absence or presence of εACA. Annexin V labeled with either Alexa fluor 488 (AF488) or Alexa fluor 568 (AF633; 1/20, Life Technologies) was added after 45 minutes. Platelet-derived plasminogen was detected using a mouse monoclonal antibody labeled in-house with either DyLight 633 or 488 (DL633 or DL488) labeling kit (Life Technologies). This antibody preferentially recognizes receptor-induced binding sites that are latent on Glu-plasminogen but become available on binding of Glu-plasminogen to cell surfaces.36,37 Plg-RKT was detected with anti–Plg-RKT mAb (7H1)28 labeled in house with DyLight 550 or 633 (DL550 or DL633) labeling kit (Life Technologies). Specificity of the antibodies was confirmed using corresponding isotype controls (supplemental Figure 1 available on the Blood Web site). A minimum of 10 000 events were collected using a Fortessa flow cytometer (Becton Dickinson) or a YETI flow cytomter (Bio-Rad). Data analysis was performed using FlowJo software (Tree Star Inc).
Fluorescent confocal microscopy
Washed platelets at 0.5 × 108 platelets/mL in HEPES buffer, pH 7.45 (1% BSA), were adhered to µ-Ibidi VI0.4–coated with 0.6 µg equine tendon type I collagen (American Biochemical Pharmaceuticals) ± 3 pmol thrombin or 0.45 nmol TRAP-6. In some experiments anti-plasminogen mAb-DL633 or anti–Plg-RKT mAb-DL550 were included during stimulation. After stimulation, annexin V-AF488 (1/20 dilution; Life Technologies) and 2 mM CaCl2 were added. For time course analysis, platelets were added directly to the coated surface with annexin V-AF568 (1/20 dilution; Life Technologies), anti-plasminogen mAb -DL633, P-selectin AF488 (Biolegend), and 2 mM CaCl2. Images were recorded on Zeiss 710 laser scanning confocal microscope with a 63 × 1.40 oil immersion objective using Zeiss Zen 2012 software.
Turbidimetric fibrinolysis assay
Purified human plasminogen-free fibrinogen (2.4 μM, Enzyme Research Laboratories), ± glu-plasminogen (0.24 μM, Enzyme Research Laboratories) ± 2.5 × 108 washed platelets/mL, in HEPES buffer, pH 7.45, was added in triplicate to 96-well polystyrene plates. Clotting was initiated by thrombin (0.25 U/mL) and CaCl2 (5 mM), and turbidity was monitored every minute at 340 nm for 30 minutes at 37°C in a FLX-800 plate reader (Biotek Instruments). After 30-minute polymerization, 1 nM tPA (Genentech) was overlaid onto each clot, and turbidity was monitored for 5 hours.
Fluorescent imaging of clot lysis
Clots were prepared as above with the inclusion of plasminogen-free fibrinogen labeled with DyLight 488 (0.25 μM). The clots were formed in µ-Ibidi VI0.4 chamber slides and incubated at 37°C for 30 minutes. After this time, 75 nM tPA was added to the edge of the clots, and the lysis front was monitored on a UVP Biospectrum 810 imaging system taking an image every 15 minutes for 18 hours.
Western blotting
Human platelets (1 × 107) were lysed (L) and separated by ultracentrifugation into soluble protein (S) and membrane (M) fractions. The fractions were run on 4% to 12% NuPage gels followed by western blotting with anti–Plg-RKT mAb.
Tail bleeding assay
Mice were anesthetized with isoflurane in prone position. A distal 4-mm segment of the tail was amputated, and the tail was immediately immersed in isotonic saline prewarmed to 37°C. The position of the tail was vertical, with the tip positioned about 2 cm below the body horizon, and the time until bleeding ceased was monitored.
Statistical analysis
Statistical analysis was performed in GraphPad Prism 5.04 (GraphPad Software LLC) using 1-way analysis of variance with Bonferroni post hoc test or an unpaired Student t test. P < .05 was considered significant. Results are represented by the mean ± standard error of the mean (SEM).
Results
Platelet-derived plasminogen is retained on the activated membrane
To determine whether platelets contain a pool of plasminogen, we quantified antigen levels in the supernatant of stimulated platelets using an ELISA. The supernatants from unstimulated platelets and platelets stimulated with collagen + thrombin contained similar levels of plasminogen (0.08 vs 0.07 nmol/108 platelets, respectively; Figure 1A). Blocking lysine-dependent plasminogen binding to the platelet surface by isolating platelets in the presence of the lysine analog εACA resulted in a 4.7-fold increase in plasminogen content to 0.33 nmol/108 platelets in the supernatant fraction (Figure 1A; P < .05). These data indicate that platelet-derived plasminogen is released from platelets on stimulation and is then retained on the activated membrane. Similarly, flow cytometry revealed a 3.7-fold increase in platelet-associated plasminogen on stimulation with thrombin + CVX compared with unstimulated platelets (Figure 1B-C; P < .01). Thrombin alone resulted in the same level of platelet-associated plasminogen as that stimulated by thrombin + CVX (median fluorescence intensity [MFI]: 602.8 ± 162.9 vs 605.0 ± 102.1). Stimulation with TRAP-6 and CVX resulted in a 2.3-fold increase in plasminogen exposure compared with unstimulated platelets. Platelet-associated plasminogen was apparent in both PS-positive and PS-negative platelets (Figure 1D); however, the vast majority in the thrombin + CVX group were PS-exposing platelets. Inclusion of εACA during platelet stimulation with thrombin + CVX significantly reduced plasminogen exposure (MFI: 159.0 ± 58.8 vs 605.0 ± 102.1; Figure 1B-C; P > .01), consistent with the ELISA data.

Plasminogen is released from activated platelets and retained on the stimulated membrane. (A) Isolated human platelets were stimulated with collagen (20 µg/mL) and thrombin (100 nM) in the absence (hatched bars) or presence of εACA (200 mM, solid bars) for 30 minutes. Plasminogen was detected in the supernatant by ELISA. (B-D) Platelets (2 × 108 platelets/mL) were stimulated ± CVX (100 ng/mL) with TRAP-6 (15 μM) or thrombin (100 nM) ± εACA. Annexin V-AF488 and anti-plasminogen antibody-DL633 were included to detected PS exposure and platelet-derived plasminogen, respectively. (B) Representative flow cytometry curves. Data are presented as mean ± SEM for (C) MFI for platelet-derived plasminogen exposure and (D) PS exposure in plasminogen-positive platelets. PS-negative platelets are indicated by hatched bars and PS positive by open bars. Color coding as in panel C. *P < .05, **P < .01; n ≥ 3.
Plasminogen is released from activated platelets and retained on the stimulated membrane. (A) Isolated human platelets were stimulated with collagen (20 µg/mL) and thrombin (100 nM) in the absence (hatched bars) or presence of εACA (200 mM, solid bars) for 30 minutes. Plasminogen was detected in the supernatant by ELISA. (B-D) Platelets (2 × 108 platelets/mL) were stimulated ± CVX (100 ng/mL) with TRAP-6 (15 μM) or thrombin (100 nM) ± εACA. Annexin V-AF488 and anti-plasminogen antibody-DL633 were included to detected PS exposure and platelet-derived plasminogen, respectively. (B) Representative flow cytometry curves. Data are presented as mean ± SEM for (C) MFI for platelet-derived plasminogen exposure and (D) PS exposure in plasminogen-positive platelets. PS-negative platelets are indicated by hatched bars and PS positive by open bars. Color coding as in panel C. *P < .05, **P < .01; n ≥ 3.
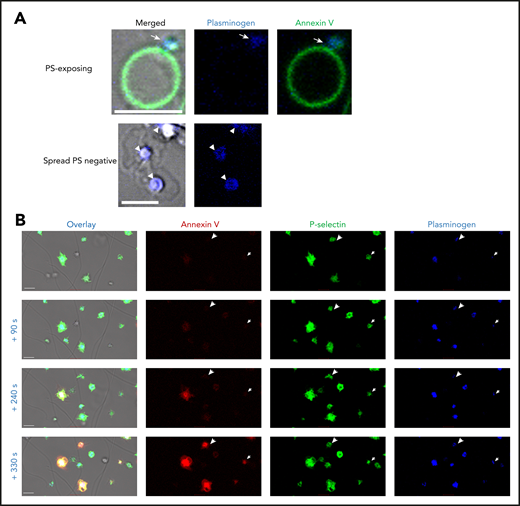
We investigated the localization of platelet-derived plasminogen on the platelet surface using confocal microscopy. In PS-exposing platelets (Annexin V positive), plasminogen was detected in the cap or platelet body (Figure 2A). Plasminogen was located centrally over the granulomere in spread platelets. P-selectin is commonly used as a marker of platelet α-granule release. P-selectin and platelet-derived plasminogen were simultaneously detected on the surface of platelets on a thrombin- and collagen-coated surface (Figure 2B). Interestingly, exposure of P-selectin and plasminogen on the activated platelet-membrane was observed before detectable PS. These data indicate that complete PS is not required for plasminogen release. Furthermore, concurrent exposure with P-selectin is consistent with plasminogen retention in α-granules.
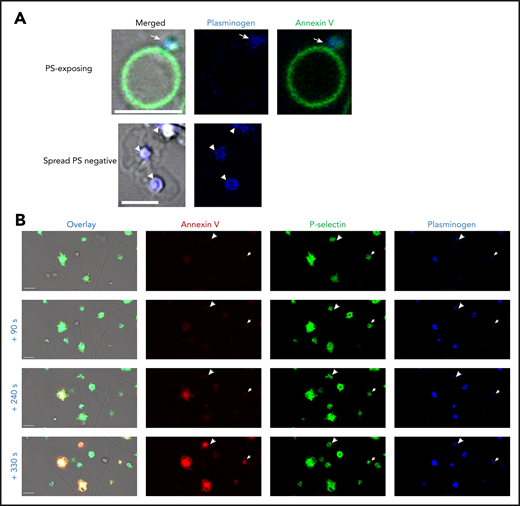
Platelet-derived plasminogen is retained in a distinct pattern on activated platelets. Platelets (0.5 × 108 platelets/mL) were stimulated on a collagen (0.6 µg)- and thrombin (3 pmol)-coated surface stained with (A) Annexin V-AF488 and anti-plasminogen antibody DL633. Arrow indicates plasminogen in the platelet cap. Arrowheads indicate plasminogen over the granulomere. (B) Time course of platelet activation. Platelets are labeled with Annexin V-AF568, AF488 P-selectin antibody, and anti-plasminogen antibody DL633. Arrowheads indicate the single platelets at different time points. Scale bars, 5 μm; representative images of n ≥ 3.
Platelet-derived plasminogen is retained in a distinct pattern on activated platelets. Platelets (0.5 × 108 platelets/mL) were stimulated on a collagen (0.6 µg)- and thrombin (3 pmol)-coated surface stained with (A) Annexin V-AF488 and anti-plasminogen antibody DL633. Arrow indicates plasminogen in the platelet cap. Arrowheads indicate plasminogen over the granulomere. (B) Time course of platelet activation. Platelets are labeled with Annexin V-AF568, AF488 P-selectin antibody, and anti-plasminogen antibody DL633. Arrowheads indicate the single platelets at different time points. Scale bars, 5 μm; representative images of n ≥ 3.
Platelet-derived plasminogen is functional in fibrinolysis
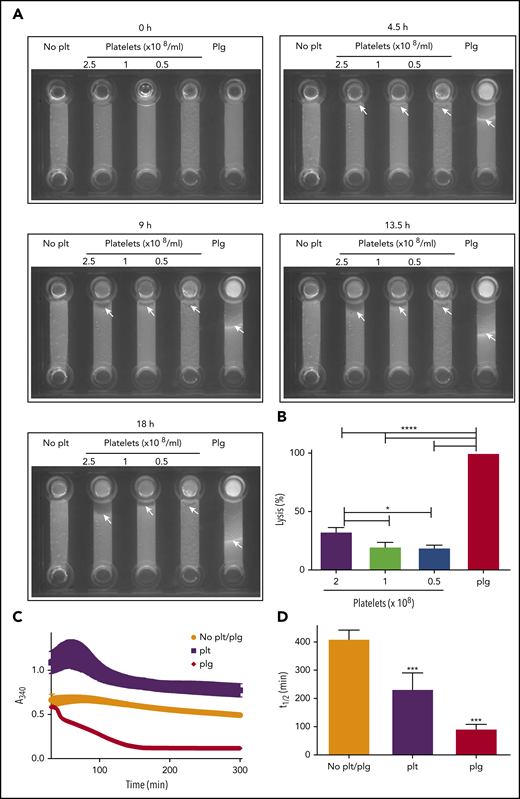
We previously reported that the activated platelet membrane is a site for plasmin generation.12 To determine whether cell surface–generated plasmin is sufficient to drive fibrinolysis, we formed purified plasminogen-free fibrinogen clots in the absence and presence of platelets. Incorporation of fluorescently labeled fibrinogen allowed the progression of lysis to be monitored after addition of tPA to the top of the chamber. A distinct lysis front was visible in a control sample containing purified plasminogen (Figure 3A; supplemental Video 1). In the absence of platelets, no fibrinolytic activity was observed. However, on inclusion of platelets, fibrinolysis was initiated. Platelets (2.5 × 108 platelets/mL) lysed 32.8 ± 3.6% of the distance lysed by the control sample containing plasminogen (Figure 3A-B; supplemental Video 1). Consistent with these observations, in turbidimetric clot lysis assays, lysis was observed on inclusion of platelets in plasminogen-deficient clots. Platelets induced lysis with a time to 50% lysis of 233.3 ± 57.2 minutes; P > .05; Figure 3C-D). Addition of purified plasminogen (0.24 μM) in the absence of platelets resulted in lysis with a 50% lysis time of 93 ± 15.5 minutes. The plasminogen concentration in 2.5 × 108 platelets is estimated to be 0.83 μM based on the ELISA data (Figure 1A). Despite this, the rate of lysis in the presence of platelets 3.6-fold slower than the plasminogen control (0.24 μM).
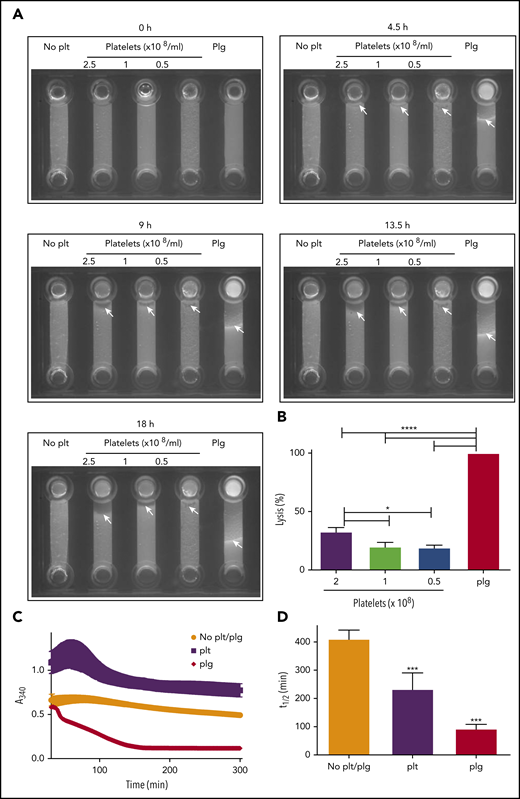
Platelets can drive fibrinolysis via a plasmin-mediated mechanism. (A-B) Purified clots were formed from plasminogen-free fibrinogen (2.4 μM) and DL488 plasminogen-free fibrinogen (0.25 μM) in the presence or absence of glu-plasminogen (0.24 μM) and in the presence or absence of the indicated concentration of platelets. Clotting was initiated by thrombin (0.25 U/mL) and CaCl2 (5 mM), and clots were allowed to form for 30 minutes before addition of 75 nM tPA to the edge of the clots. Lysis was monitored by imaging every 15 minutes for 18 hours. (B) The distance lysed at 18 hours as a percentage of the plasminogen control (set to 100%). *P < .05 and **** P < .0001 compared with plasminogen control clots. (C-D) Purified clots were formed as above with or without 2.5 × 108 platelets/mL in the absence of DL488 fibrinogen. Turbidity was monitored every minute at 340 nm for 30 minutes at 37°C in a FLX-800 plate reader (Biotek Instruments). After 30-minute polymerization, 1 nM tPA (Genentech) was overlaid onto each clot, and turbidity was subsequently monitored every minute for 5 hours. (D) Fifty percent lysis times. ***P < .01 compared with no plasminogen control clots. Data are expressed as mean ± SEM, n ≥ 3.
Platelets can drive fibrinolysis via a plasmin-mediated mechanism. (A-B) Purified clots were formed from plasminogen-free fibrinogen (2.4 μM) and DL488 plasminogen-free fibrinogen (0.25 μM) in the presence or absence of glu-plasminogen (0.24 μM) and in the presence or absence of the indicated concentration of platelets. Clotting was initiated by thrombin (0.25 U/mL) and CaCl2 (5 mM), and clots were allowed to form for 30 minutes before addition of 75 nM tPA to the edge of the clots. Lysis was monitored by imaging every 15 minutes for 18 hours. (B) The distance lysed at 18 hours as a percentage of the plasminogen control (set to 100%). *P < .05 and **** P < .0001 compared with plasminogen control clots. (C-D) Purified clots were formed as above with or without 2.5 × 108 platelets/mL in the absence of DL488 fibrinogen. Turbidity was monitored every minute at 340 nm for 30 minutes at 37°C in a FLX-800 plate reader (Biotek Instruments). After 30-minute polymerization, 1 nM tPA (Genentech) was overlaid onto each clot, and turbidity was subsequently monitored every minute for 5 hours. (D) Fifty percent lysis times. ***P < .01 compared with no plasminogen control clots. Data are expressed as mean ± SEM, n ≥ 3.
Plg-RKT is exposed on PS-exposing and spread platelets
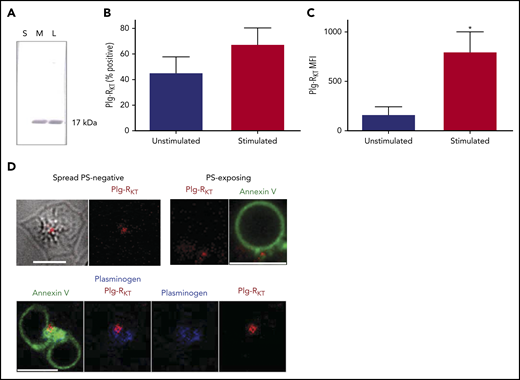
We investigated the localization of Plg-RKT in platelets. Western blots were performed on platelet fractions using an anti–Plg-RKT monoclonal antibody with a specificity for the 9 C-terminal amino acids of Plg-RKT.29 A 17-kDa band, corresponding to Plg-RKT, was identified in total platelet lysates and in the membrane fraction but was not detectable in the soluble fraction (Figure 4A). Flow cytometric analysis confirmed the presence of Plg-RKT on the platelet membrane, with a similar number of positive platelets observed before and after stimulation with thrombin and convulxin (Figure 4B). In contrast, the MFI value increased 4.8-fold on stimulation with thrombin + CVX (Figure 4C), indicative of the release of the transmembrane receptor Plg-RKT from platelet pools and its retention on the platelet surface. The localization of Plg-RKT on the platelet surface was visualized using confocal microscopy. The receptor localized to the center of spread PS-negative platelets and in the cap region of PS-exposing platelets (Figure 4D). Platelet-derived plasminogen and Plg-RKT were detected in the same locale on the different platelet subtypes.

Platelets express the transmembrane Plg-RKT. (A) Fractions of platelets (1 × 107), lysate (L), soluble (S), and membrane (M) were run on 4% to 12% NuPAGE gels followed by western blotting with anti–Plg-RKT mAb (7H1) fractions. Representative image of n = 4. (B-C) Platelets (2 × 108 platelets/mL) were either unstimulated or stimulated with CVX (100 ng/mL) plus thrombin (100 nM) in the presence of anti–Plg-RKT mAb-DL550. Data are presented as mean ± SEM for (B) percentage platelets positive for Plg-RKT and (C) MFI. *P < .05 vs unstimulated, n = 3. (D) Platelets (0.5 × 108 platelets/mL) stimulated on a collagen (0.6 µg) and thrombin (3 pmol) coated surface for 45 minutes were stained with Annexin V-AF488, anti-plasminogen mAb-DL633, and anti-Plg-RKT mAb-DL550. Scale bars, 5 μm; representative images of n = 4.
Platelets express the transmembrane Plg-RKT. (A) Fractions of platelets (1 × 107), lysate (L), soluble (S), and membrane (M) were run on 4% to 12% NuPAGE gels followed by western blotting with anti–Plg-RKT mAb (7H1) fractions. Representative image of n = 4. (B-C) Platelets (2 × 108 platelets/mL) were either unstimulated or stimulated with CVX (100 ng/mL) plus thrombin (100 nM) in the presence of anti–Plg-RKT mAb-DL550. Data are presented as mean ± SEM for (B) percentage platelets positive for Plg-RKT and (C) MFI. *P < .05 vs unstimulated, n = 3. (D) Platelets (0.5 × 108 platelets/mL) stimulated on a collagen (0.6 µg) and thrombin (3 pmol) coated surface for 45 minutes were stained with Annexin V-AF488, anti-plasminogen mAb-DL633, and anti-Plg-RKT mAb-DL550. Scale bars, 5 μm; representative images of n = 4.
Plg-RKT functions to retain plasminogen on the activated platelet membrane
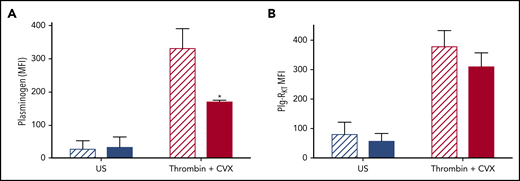
To study the role of Plg-RKT in retention of plasminogen on the platelet surface, platelets were isolated from Plg‐RKT−/− mice and plasminogen retention quantified by flow cytometry. Tail bleeding times in Plg-RKT−/− mice were not different from Plg-RKT+/+ mice, suggesting normal platelet function in these animals (supplemental Figure 2). Consistent with the human data, murine Plg-RKT+/+ platelets retained platelet-derived plasminogen on their surfaces, which was increased 12.2-fold on stimulation with thrombin + CVX. Plasminogen retention was significantly attenuated in Plg‐RKT−/− mice (Figure 5A), whereas no genotype effect was noted in unstimulated platelets. This indicates that approximately half of the pool of plasminogen that is exposed on stimulation is retained via Plg‐RKT. Exposure of Plg‐RKT was increased on stimulation of platelets isolated from Plg-RKT+/+ mice, consistent with the data from human platelets (Figure 5B). Deficiency of plasminogen did not significantly affect exposure of Plg‐RKT after stimulation, indicating that plasminogen is not required to induce exposure of this receptor (Figure 5B).
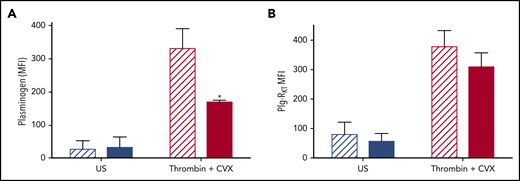
Plg-RKT −/− platelets retain reduced membrane-bound plasminogen. Washed platelets (2 × 108 platelets/mL) in modified Tyrodes buffer (pH 7.45) from Plg-RKT+/+ (hatched bars) or Plg-RKT−/− mice (solid bars) were left unstimulated or stimulated in the presence of 2 mM CaCl2 with 100 ng/mL CVX + 100 nM thrombin. Annexin V-AF633 with 2 mM CaCl2 was added after 45 minutes. Platelet-derived plasminogen was detected using anti-plasminogen DL488 antibody. A minimum of 10 000 events was collected using a Fortessa flow cytometer. *P < .05 compared with Plg-RKT+/+ stimulated platelets. Data are expressed as mean ± SEM, n ≥ 4.
Plg-RKT −/− platelets retain reduced membrane-bound plasminogen. Washed platelets (2 × 108 platelets/mL) in modified Tyrodes buffer (pH 7.45) from Plg-RKT+/+ (hatched bars) or Plg-RKT−/− mice (solid bars) were left unstimulated or stimulated in the presence of 2 mM CaCl2 with 100 ng/mL CVX + 100 nM thrombin. Annexin V-AF633 with 2 mM CaCl2 was added after 45 minutes. Platelet-derived plasminogen was detected using anti-plasminogen DL488 antibody. A minimum of 10 000 events was collected using a Fortessa flow cytometer. *P < .05 compared with Plg-RKT+/+ stimulated platelets. Data are expressed as mean ± SEM, n ≥ 4.
Discussion
It is now established that platelets can bind exogenous plasminogen,11,12,19,38 but the potential contribution of platelets as a source of plasminogen has not been explored. Here we demonstrate that platelets harbor a discrete pool of plasminogen that is secreted after platelet activation. Plasminogen localized to the platelet cap in PS-exposing platelets and centrally in spread platelets. Importantly, we demonstrate for the first time that this platelet-derived plasminogen facilitates fibrinolysis in the absence of exogenous plasminogen. This highlights a potential function of platelets to drive local fibrinolysis. Platelet-derived plasminogen is retained on the activated platelet membrane via a lysine-dependent manner. Genetic ablation of the transmembrane receptor, Plg-RKT, attenuates binding of plasminogen by approximately 50% highlighting the importance of this receptor in retention of plasminogen on the activated platelet membrane.
Multiple receptors for plasminogen have been described in various cell types, but not all cell types express the same receptors.26 A common feature of these receptors is that they bind plasminogen via its lysine binding sites within the kringle domains.39 Glu-plasminogen has a relatively closed structure and crystallographic studies indicate that only the lysine binding domain in kringle one is accessible for initial binding.40 Binding to lysine induces a more open easily activated conformation,41 thus facilitating further attachment to surfaces augmenting activation. Furthermore, binding of plasminogen to cellular surfaces protects plasmin generated on the surface from inhibition by α2AP.23-25 Inclusion of the lysine analog, εACA, to block lysine-dependent binding completely abolishes retention of platelet-derived plasminogen, indicative of a lysine-dependent mechanism. Plg-RKT is the first integral transmembrane receptor described to be synthesized with a C-terminal lysine. Plg-RKT promotes plasminogen activation, cell migration, recruitment, and polarization of macrophages.29,31 Here, we establish that Plg-RKT is exposed on stimulation of platelets and accounts for approximately half of the pool of platelet-derived plasminogen retained on platelets. Tail bleeding times were normal in these mice indicating normal hemostatic platelet function. If this receptor has a role in the intracellular trafficking of plasminogen or indeed uptake of the protein this could contribute to the reduction of plasminogen. Nonetheless, this data suggests that a deficiency in this receptor could therefore significantly impact retention of plasminogen altering the fibrinolytic potential on the platelet surface. The receptor and plasminogen were visualized in the cap of PS-exposing platelets and centrally in spread platelets. This is consistent with our previous observation of the localization of binding of plasma-derived plasminogen in both subpopulations of platelets.12
We previously demonstrated that a binding of exogenous plasminogen primarily occurs via a fibrin- and αIIbβ3-dependent mechanism.12 Consistent with a fibrin-dependent mechanism, stimulation with TRAP-6 did not induce plasminogen retention to the same level as that observed with thrombin stimulation. TRAP-6 is a PAR-1 agonist, but unlike thrombin, does not cleave fibrinogen to fibrin, thereby curbing the number of available binding sites. Fibrin(ogen) binding and polymerization stabilizes the platelet cap via an αIIbβ3-dependent mechanism14 . Clustering of fibrin(ogen) and αIIbβ3 within the cap is required for incorporation of PS-exposing platelets into the growing thrombus, with the cap serving as the point of contact with platelet aggregates.14 Concentration of plasminogen in the cap could provide a focal point for directing fibrinolysis. Indeed, the concentration of procoagulant factors within the cap has been proposed to accelerate the clotting reaction and subsequent fibrin formation.17
Platelets were capable of driving fibrinolysis in a system free from additional plasminogen, highlighting the importance of this cellular source. Inclusion of platelets in clots at 2.5 × 108 gives rise to a plasminogen concentration of around 0.83 μM; however, the overall rate of lysis was slower compared to the plasminogen control (0.24 μM). This is perhaps not surprising considering the abundance of antifibrinolytic proteins in platelets. Active PAI-1 is present in high concentrations1,2,42 and is retained on the activated platelet membrane,2 and platelets are a source of additional inhibitors, including TAFI,43,44 α2AP45 and the transglutaminase FXIII-A.46 These data highlight the complex nature of the host of fibrinolytic proteins on the activated platelet membrane in regulating fibrinolysis. Differential release of pro- and antiangiogenic factors from activated platelets has been demonstrated47 and poses the question as to whether this phenomenon also occurs for pro- and antifibrinolytic factors.
The platelet membrane enhances activation of scuPA in a mechanism of reciprocal plasminogen activation that is independent of uPAR.19 There are conflicting reports on existence of uPAR in platelets.48,49 However, binding of uPA has been shown to bind to leukemic cell lines in the absence of uPAR.50 Indeed, activation of plasminogen by uPA can occur via a cross talk mechanism that bypasses the requirement for uPA and plasminogen to be present on the same cell surface.51 We recently demonstrated that plasminogen activation by factor XIIa (FXIIa) is enhanced in the presence of platelet-derived polyphosphate (polyP).52 Along with plasminogen, FXIIa and platelet-derived polyP accumulate on the activated platelet membrane.52 Patients with Quebec platelet disorder exhibit a bleeding diathesis, because of augmented levels of uPA within α-granules of platelets, thereby promoting aberrant degradation of α-granule proteins.34 The fibrinolytic capacity of the platelet membrane is clearly dependent on the balance and the assembly of these fibrinolytic proteins on the surface and the surrounding milieu.
Targeting the fibrinolytic potential of the platelet surface is therefore an interesting potential for thrombolytic therapies. Indeed, fusing scuPA to the platelet surface via αIIbβ3 prevents occlusion in a mouse ferric chloride model.53 This process was plasminogen dependent, and importantly, use of this fusion protein did not prolong bleeding times in animals. Previously we demonstrated that plasminogen accumulated at the base or core of thrombi in a whole blood model of thrombus formation and lysis. This was apparent both before and after visible fibrin formation.12 The thrombus architecture is heterogeneous consisting of a core of fully activated, densely packed platelets and an outer shell of less-activated, loosely associated platelets.54-58 The microenvironment of the tightly packed core of activated platelets dictates solute transport, with restricted protein diffusion evident.54,56,59 Accumulation, activation, and packing of platelets are critical to limiting plasma extravasation.60 The relatively large size of plasminogen (92 kDa) would limit the ability of plasma plasminogen to penetrate the thrombus core. Endogenous fibrinolytic activity has been observed within thrombi, which facilitates the contractile forces of platelets on fibrin during the process of clot retraction.61 Plasmin is also reported to affect platelet function by inhibition of aggregation and degradation of glycoprotein 1b.62-64 Treatment of platelets with the widely used thrombolytic Alteplase has recently been shown to interfere with clot retraction.65 Therefore, our observation that plasminogen concentrates at the thrombus core suggests that platelet-derived or associated plasminogen could drive internal remodeling of the thrombus and promote early resolution.
Our data conclusively demonstrate that platelets contain a pool of intracellular plasminogen that is retained on the surface of platelets following activation. Approximately half of the platelet-derived pool of plasminogen is anchored via the transmembrane receptor Plg-RKT. These data together with our previous publications suggest that the platelet surface acts as scaffold housing both pro- and antifibrinolytic factors, the balance of which has important implications in driving fibrinolysis. Harnessing platelets endogenous potential for plasminogen activation could provide a new avenue for novel thrombolytics.
For original data, please contact n.j.mutch@abdn.ac.uk or c.s.whyte@abdn.ac.uk.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Microscopy and Histology Core Facility and the Iain Fraser Cytometry at the University of Aberdeen for excellent advice and use of facilities. The authors thank Victoria Ploplis (University of Notre Dame, South Bend, IN) for kindly providing mice deficient in plasminogen. The visual abstract was created with BioRender.com.
This study was supported by British Heart Foundation grant PG/15/82/31721 (C.S.W. and N.J.M.), British Society for Haematology, Thrombosis UK, British Society of Haemostasis & Thrombosis Joint Studentship (G.B.M., C.S.W., and N.J.M.), University of Tabuk Studentship (M.M.J. and N.J.M.), National Institutes of Health, National Heart, Lung, and Blood Institute grants HL-081046 (L.A.M.) and HL149511 (R.J.P. and L.A.M.), and Merit Review Award I01BX003933 from the US Department of Veterans Affairs (R.J.P.).
Authorship
Contribution: C.S.W. performed the research, analyzed the data, and wrote the manuscript; G.B.M., N.B., M.M.J., and N.A.B. performed the research and analyzed data; R.J.P. and L.A.M. supervised the research and edited the manuscript; and N.J.M. supervised the research, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Nicola J. Mutch, Aberdeen Cardiovascular and Diabetes Centre, School of Medicine, Medical Sciences & Nutrition, Institute of Medical Sciences, Foresterhill, University of Aberdeen, Aberdeen AB25 2ZD, United Kingdom; e-mail: n.j.mutch@abdn.ac.uk.