

Key Points
Patients with sepsis show an acquired platelet function defect with massive loss of GPVI receptor responsiveness.
Platelet GPVI dysfunction is evident already at early disease onset and restores when patients recover.

Abstract
Glycoprotein VI (GPVI), the platelet immunoreceptor tyrosine activating motif (ITAM) receptor for collagen, plays a striking role on vascular integrity in animal models of inflammation and sepsis. Understanding ITAM-receptor signaling defects in humans suffering from sepsis may improve our understanding of the pathophysiology, especially during disease onset. In a pilot study, platelets from 15 patients with sepsis were assessed consecutively at day of admission, day 5 to 7, and the day of intensive care unit (ICU) discharge and subjected to comprehensive analyses by flow cytometry, aggregometry, and immunoblotting. Platelet function was markedly reduced in all patients. The defect was most prominent after GPVI stimulation with collagen-related peptide. In 14 of 15 patients, GPVI dysfunction was already present at time of ICU admission, considerably before the critical drop in platelet counts. Sepsis platelets failed to transduce the GPVI-mediated signal to trigger tyrosine phosphorylation of Syk kinase or LAT. GPVI deficiency was partially inducible in platelets of healthy donors through coincubation in whole blood, but not in plasma from patients with sepsis. Platelet aggregation upon GPVI stimulation increased only in those patients whose condition ameliorated. As blunted GPVI signaling occurred early at sepsis onset, this defect could be exploited as an indicator for early sepsis diagnosis, which needs to be confirmed in prospective studies.
Introduction
In sepsis, thrombocytopenia is frequent and, at least in part, considered to be a consequence of disseminated intravasal coagulation (DIC). A low platelet count is accounted for in the diagnostic sequential organ failure assessment (SOFA) score and associated with a poor prognosis.1 In addition, an increasing body of evidence suggests that platelets are also functionally altered in sepsis.2 As sepsis definitions have been specified over the last decades and patient cohorts differ substantially between distinct studies, the identified platelet defects do not provide a clear picture but are rather contradictory. Most platelet agonists like ADP or thrombin act via G protein–coupled receptors that lead to calcium influx, followed by integrin activation and release of internal granules.3 Collagen, in contrast, binds to the immunoreceptor glycoprotein VI (GPVI), which might have also binding sites for other ligands like fibrin.4,5 CLEC-2 is a related hem–immunoreceptor tyrosine activating motif (ITAM) receptor with podoplanin as the so far only identified physiological ligand.6
GPVI and CLEC-2 are essential for vascular integrity, especially at sites of inflammation.7,8 Mouse models for sepsis are well established, but typically hampered by the fact that the Fc receptor repertoire differs substantially between humans and mice, with, in part, opposing signaling cascades. Claushuis and colleagues have shown impaired immune response of GPVI−/− mice in a Klebsiella pneumoniae inhalation model, whereas depletion of CLEC-2 did not affect any responses during the induced pneumonia.9 In contrast, Rayes and colleagues could not find a role of GPVI, but for CLEC-2 in systemic inflammation and organ damage, using 2 mouse models of sepsis-intraperitoneal lipopolysaccharide and cecal ligation and puncture.10 Although mouse models and bacterial strains differ, there is a growing body of evidence that the signaling cascade shared by ITAM and hemITAM receptors is involved in septic inflammation.11
So far, there are only a few comprehensive reports on platelet function in human sepsis.12-14 Gawaz et al reported that platelets in septic patients are in a preactivated state that could lead to microthrombotic events.15,16 The observational prospective PRESS study has recently identified an increased platelet reactivity using point-of-care devices during early sepsis, but not in septic shock.17
Here, we provide results from a prospective study, where we assessed platelet function at different time points in the course of sepsis. Platelets were hyporeactive to multiple agonists, and a significant ITAM-receptor signaling defect was present already at time of intensive care unit (ICU) admission. We conclude that (hem)ITAM-based platelet function analysis could be exploited as a biomarker for early sepsis.
Methods
Patient recruitment and blood collection
The study was approved by the ethics committee of the University of Würzburg (reference: 102/17-sc) and conducted according to the Declaration of Helsinki, including its current amendments. Informed consent was provided in written form by the patient, a family member, or a legal guardian. Adult patients with sepsis (age ≥18 years) were recruited between September 2017 and May 2018 from 2 ICUs of the University Hospital Würzburg, comprising a mixed medical/surgical patient cohort. Venous blood from patients that were administered at one of the indicated ICUs was drawn through routine-supply catheters into sodium-citrate (3.2%) monovettes. Patients were included when diagnosed with sepsis according to both sepsis I and sepsis III criteria. A diagnosis of “septic shock” was assigned when at least one of the following criteria was additionally present: (1) vasopressors essential for the maintenance of a mean arterial pressure above 65 mmHg; (2) serum-lactate ≥2 mmol/L (supplemental Figure 1, available on the Blood Web site). Patients were excluded due to pregnancy or an underlying hematooncological disorder. Blood of adult healthy volunteers, self-reporting of not having taken antiplatelet drugs for the last 10 days, was collected by venous puncture.
Additional methods on flow cytometry, platelet function tests, immunoblot analysis, enzyme-linked immunosorbent assay (ELISA), bacteria, and the used statistical methods can be found in the supplement data.
Results
Fifteen patients diagnosed with sepsis according to sepsis I18 and sepsis III19 criteria were recruited between September 2017 and May 2018 from 2 ICUs of the University Hospital Würzburg (details in supplemental Figure 1 and supplemental Table 1) and followed up during disease progression until the day of discharge from the ICU. Most patients were included very early during disease progression. Eight of 15 patients were transferred to the ICU within 12 hours, 12 of 15 within 24 hours, and 15 of 15 within 36 hours after presentation at the hospital with sepsis symptoms. Blood withdrawal was performed in 5 of 15 patients within 24 hours, in 13 of 15 patients within 48 hours, and in 15 of 15 patients within 72 hours after hospital presentation.
Median age was 70 years with a range of 19 to 84 years. The focus of infection, identified pathogens, and applied therapies were diverse: only 1 patient was under clopidogrel therapy, and 1 patient received platelet transfusions of in total 1 platelet concentrate before the follow-up analysis (Tables 1 and,2). Two patients had previous surgery (ID 32: laparoscopic cholecystectomy; ID 33: osteosynthesis + exploratory laparotomy). As a control group, 19 healthy individuals (median age, 32 years; range, 22 to 61) were included that had not taken platelet function-relevant medication (COX inhibitors or thienopyridines) for 10 days prior to blood sampling.
Elevated platelet biogenesis during sepsis progression
On ICU admission, median platelet count in our cohort was 167/nL (IQR, 122.5; 231.5). Eight patients had a normal platelet count; 5 patients were thrombocytopenic (<150/nL), and 2 patients had counts >350/nL. In 6 of the 10 patients with initially regular or increased platelets, the counts decreased below the lower reference value during the first week on ICU. Two patients did not develop thrombocytopenia during the course of disease. Until day of discharge, platelet counts had normalized in every patient (Figure 1A). In order to determine whether sepsis had an impact on platelet production, we analyzed the fraction of thiazole orange (TO)-positive, reticulated platelets,20 which increased during the first 5 days in ICU (supplemental Figure 2; Figure 1B). Thrombopoietin (TPO) plasma levels were markedly elevated in patients with sepsis compared with the reference range (0-168 pg/mL), implying that the regulatory feedback loop to drive thrombopoiesis in response to low platelet counts is functional during the course of disease (Figure 1C). Finally, mean platelet volume, which is an independent marker of newly formed platelets, increased significantly until time point II (Figure 1D).

Elevated platelet biogenesis during sepsis progression. Characteristics of patients with sepsis (S) are displayed at the following time points: I: admission day; II: day 5 to 7; and III: day of ICU discharge. (A) Platelet count. (B) Reticulated platelets indicated as TO-positive fraction were assessed by flow cytometry. (C) TPO levels in sepsis plasma were determined by ELISA. (D) Mean platelet volume. (E) Main platelet receptor and integrin expression were analyzed by flow cytometry in patients and corresponding healthy controls (HC). (F-I) Platelet (pre-)activation due to P-selectin exposure (CD62P) (F-G) and integrin αIIbβ3 activation (H-I) were assessed in whole blood measured by flow cytometry. Representative curves are shown in panels F and H. (A,D) Reference ranges are shown as dashed lines. (A-I) Graphs show median ± IQR. Differences were analyzed using Wilcoxon matched-pairs signed rank test (A,C,D). Kolmogorov-Smirnov test (E). Kruskal-Wallis test (B, G, I). n.s., nonsignificant. *P < .05; **P < .01; ***P < .001; ****P < .0001. APC, allophycocyanin; FITC, fluorescein isothiocyanate; GEO-MFI, geometric mean fluorescence intensity; MPV, mean platelet volume; PLTs, platelets.
Elevated platelet biogenesis during sepsis progression. Characteristics of patients with sepsis (S) are displayed at the following time points: I: admission day; II: day 5 to 7; and III: day of ICU discharge. (A) Platelet count. (B) Reticulated platelets indicated as TO-positive fraction were assessed by flow cytometry. (C) TPO levels in sepsis plasma were determined by ELISA. (D) Mean platelet volume. (E) Main platelet receptor and integrin expression were analyzed by flow cytometry in patients and corresponding healthy controls (HC). (F-I) Platelet (pre-)activation due to P-selectin exposure (CD62P) (F-G) and integrin αIIbβ3 activation (H-I) were assessed in whole blood measured by flow cytometry. Representative curves are shown in panels F and H. (A,D) Reference ranges are shown as dashed lines. (A-I) Graphs show median ± IQR. Differences were analyzed using Wilcoxon matched-pairs signed rank test (A,C,D). Kolmogorov-Smirnov test (E). Kruskal-Wallis test (B, G, I). n.s., nonsignificant. *P < .05; **P < .01; ***P < .001; ****P < .0001. APC, allophycocyanin; FITC, fluorescein isothiocyanate; GEO-MFI, geometric mean fluorescence intensity; MPV, mean platelet volume; PLTs, platelets.
Expression levels of the main platelet surface receptors for von Willebrand factor (GPIb/IX) and fibrinogen (GPIIb/IIIa), as well as integrin β1, remained unchanged (Figure 1E). A drop in platelet count could also be due to an increased consumption, that is, in response to ectopic activation of platelets in circulation. We addressed this flow cytometrically by staining resting platelets for activated integrin αIIbβ3 (PAC-1 antibody binding) or P-selectin (CD62P neo-exposition as a marker for α-granule release). Unexpectedly, none of these markers was elevated on resting platelets from patients with sepsis when compared with controls (Figure 1F-I). Platelet preactivation was also not detected in patients diagnosed with disseminated intravascular coagulation (DIC), according to an International Society on Thrombosis and Haemostasis DIC score of 5 or more (data not shown), although these patients usually suffer from severe microthrombotic events in conjunction with increased platelet consumption.
Taken together, our data show that platelets in septic patients are not preactivated, whereas thrombopoiesis is stimulated in response to a lowered platelet count.
Platelets in sepsis are hyporeactive
Although platelet function is readily measured in specific point-of-care devices, preanalytical requirements define minimal values for platelet count and hematocrit that are often not achieved in patients with sepsis. We thus decided to perform a comprehensive platelet function analysis, comprising flow cytometry and light transmission aggregometry, as recommended for patients with platelet function defects.21,22 We incubated platelets with a synthetic crosslinked collagen-related peptide (CRP-XL) or the snake-venom convulxin to stimulate GPVI/ITAM signaling as well as with ADP, which stimulates the purinergic receptors P2Y12 and P2Y1.
Stimulation of control platelets with these agonists led to integrin activation as well as α-granule release as shown by flow cytometry (Figure 2A-F). At time I, 10 of 15 patients showed markedly decreased platelet integrin activation upon stimulation with 5 µM ADP (Figure 2A). In contrast, α-degranulation was significantly reduced in only 8 of 15 patients compared with controls (Figure 2B). When 0.01 µg/mL CRP-XL was used, almost all patients (14/15) revealed a markedly reduced response for both readouts (PAC-1 binding or CD62P exposure) (Figure 2C-D), suggesting that the response to this agonist is more severely affected. Although integrin GPIIb/IIIa activation was completely abrogated, we found that in some sepsis patients a platelet subpopulation became partially CD62P positive (see also histograms in supplemental Figure 3). When platelets were stimulated with 0.01 µg/mL convulxin, an impaired activation response was still evident, albeit to a lesser extent compared with CRP-XL (Figure 2E-F). This can be explained by the multivalent binding properties of convulxin, leading to increased GPVI clustering. Moreover, convulxin has been reported to interact with GPIb, which might modulate the signal strength compared with CRP-XL.23 Interestingly, this hyporeactivity was observed before platelet counts decreased under 150/nL, suggesting that platelets become dysfunctional during early sepsis progression. Of note, CD62P exposure was less affected as compared with integrin αIIbβ3 activation in response to all tested platelet agonists.
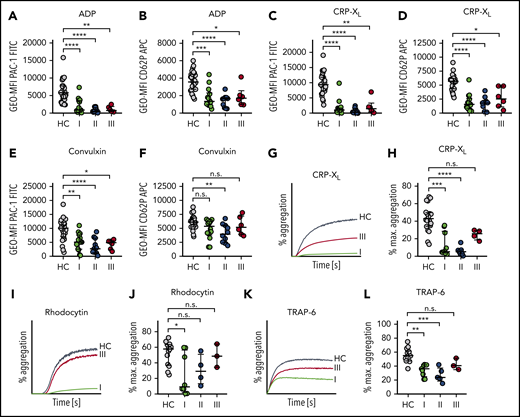
Sepsis platelets are hyporeactive. Platelet reactivity assays are shown at the following time points: I: admission day; II: day 5 to 7; and III: day of ICU discharge. (A-F) Platelet integrin activation (A,C,E) and P-selectin exposure (B,D,F) were measured upon stimulation with ADP (5 µM) (A-B), CRP-XL (0.01 µg/mL) (C-D), or convulxin (0.01 µg/mL) (E-F) in whole blood by flow cytometry. (G-L) Light transmission aggregometry was performed using washed platelets (500 000/µL). Maximum (max.) aggregation is depicted upon stimulation with CRP-XL (0.1 µg/mL) (G-H), rhodocytin (10 nM) (I-J), or TRAP-6 (10 µM) (K-L). Representative curves are shown in panels G, I, and K. CRP-XL samples were measured for 5 minutes, and rhodocytin or TRAP-6 samples for 10 minutes. Graphs show median ± IQR. Differences were analyzed by Kruskal-Wallis test (A-L). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Sepsis platelets are hyporeactive. Platelet reactivity assays are shown at the following time points: I: admission day; II: day 5 to 7; and III: day of ICU discharge. (A-F) Platelet integrin activation (A,C,E) and P-selectin exposure (B,D,F) were measured upon stimulation with ADP (5 µM) (A-B), CRP-XL (0.01 µg/mL) (C-D), or convulxin (0.01 µg/mL) (E-F) in whole blood by flow cytometry. (G-L) Light transmission aggregometry was performed using washed platelets (500 000/µL). Maximum (max.) aggregation is depicted upon stimulation with CRP-XL (0.1 µg/mL) (G-H), rhodocytin (10 nM) (I-J), or TRAP-6 (10 µM) (K-L). Representative curves are shown in panels G, I, and K. CRP-XL samples were measured for 5 minutes, and rhodocytin or TRAP-6 samples for 10 minutes. Graphs show median ± IQR. Differences were analyzed by Kruskal-Wallis test (A-L). *P < .05; **P < .01; ***P < .001; ****P < .0001.
For 8 of 15 patients, the obtained biomaterial was sufficient to also perform light transmission aggregometry. Platelet aggregation was markedly reduced during disease progression in all tested sepsis patients in response to CRP-XL when compared with controls. At time III (discharge from ICU), the maximal platelet aggregation rate in response to 0.1 µg/mL CRP-XL partially had recovered, although the difference between time II and III was not statistically significant (Figure 2G-H).
An abolished aggregation response was also detected in 5 of 8 patients at time I, when platelets were stimulated with 10 nM of CLEC-2 agonist rhodocytin, whereas platelets of 3 patients reacted normally. Maximum aggregation upon rhodocytin treatment had completely recovered at day of discharge (time III) (Figure 2I-J). Stimulation with 10 µM of PAR-1 receptor agonist TRAP-6 led to a moderate platelet aggregation of 21% at day of ICU admission (mean, 34%), indicating a remaining platelet aggregation potential under septic conditions (Figure 2K-L). Taken together, our results demonstrate that during sepsis, platelets are overall less responsive to agonists, but show a selective dysfunction toward the collagen receptor GPVI. This dysfunction recovered when the patient’s condition ameliorated.
Increased GPVI ectodomain shedding in patients with sepsis
Next, we analyzed whether the impaired platelet function was caused by altered receptor expression. Although CLEC-2 levels were unaltered, GPVI surface expression was partly reduced compared with controls (Figure 3A-B). Immunoblot analysis of platelet lysates confirmed that GPVI was slightly reduced compared with controls (Figure 3C). Protein levels were further decreased after platelet activation with CRP-XL. In line with this, plasma levels of soluble GPVI ectodomain were not markedly elevated until day 5 to 7 (time point II) compared with healthy individuals (Figure 3D).

Sepsis platelets show increased GPVI ectodomain shedding. Assays display the following time points: I: admission day; II: day 5 to 7; and III: day of ICU discharge. (A-B) CLEC-2 (A) and GPVI (B) expression on platelet surface were analyzed by flow cytometry. (C) GPVI expression studied by western blotting using JAQ-1 antibody. Platelet lysis was performed 60 minutes after stimulation with CRP-XL (C) (0.1 µg/mL) or under resting conditions (−). (D) ELISA for soluble GPVI ectodomain (sGPVI) was performed with plasma of sepsis patients and corresponding controls. Graphs show median ± IQR. Differences were analyzed by Kolmogorov-Smirnov test (A) or Kruskal-Wallis test (B,D). *P < .05; **P < .01. AU, arbitrary units; α-GAPDH, α-glyceraldehyde-3-phosphate dehydrogenase.
Sepsis platelets show increased GPVI ectodomain shedding. Assays display the following time points: I: admission day; II: day 5 to 7; and III: day of ICU discharge. (A-B) CLEC-2 (A) and GPVI (B) expression on platelet surface were analyzed by flow cytometry. (C) GPVI expression studied by western blotting using JAQ-1 antibody. Platelet lysis was performed 60 minutes after stimulation with CRP-XL (C) (0.1 µg/mL) or under resting conditions (−). (D) ELISA for soluble GPVI ectodomain (sGPVI) was performed with plasma of sepsis patients and corresponding controls. Graphs show median ± IQR. Differences were analyzed by Kolmogorov-Smirnov test (A) or Kruskal-Wallis test (B,D). *P < .05; **P < .01. AU, arbitrary units; α-GAPDH, α-glyceraldehyde-3-phosphate dehydrogenase.
Our results thus imply that the defective response to GPVI signaling is not due to ectodomain shedding and that soluble GPVI is not a suitable biomarker for early sepsis.
Platelets from patients with sepsis show impaired (hem)ITAM signaling
Next, we analyzed the GPVI/CLEC-2 signaling cascades in patients with sepsis in more detail. The global pattern of proteins that are constitutively phosphorylated in resting platelets was without any obvious difference between sepsis patients and controls (Figure 4A-B). Upon stimulation with 0.1 µg/mL CRP-XL or 10 nM rhodocytin, controls showed an induction of additional tyrosine phosphorylation, which was absent in patients with sepsis. This difference was most evident for a band of ∼72 kDa, known to reflect Syk (Figure 4A-C; supplemental Figure 4). Using phospho-epitope–specific antibodies directed against pSyk (525/526) or pLat (191), we could confirm that the ITAM-signaling cascade downstream of GPVI is markedly affected in platelets from patients with sepsis (Figure 4D-F).

Sepsis platelets show impaired (hem)ITAM signaling. Washed platelets (500 000/µL) of healthy controls (HC) and patients with sepsis (S) were stimulated with CRP-XL (C) and rhodocytin (R). Lysis was done at the following time points: C: 5 minutes; R: 10 minutes. I: admission day; II: day 5 to 7; and III: day of ICU discharge. (A-C) Western blot analysis of platelet lysates. Staining was performed with anti-phospho-tyrosine antibody 4G10. Band intensity (72 kDa) was set in relation to signal intensity of Syk or GAPDH in AU. Representative blots are shown in panels A (time point I) and B (time point III). (D-F) Phosphorylation of signaling peptides Syk and Lat was investigated by immunoblot. One representative blot is shown in panel D. (G-J) Phosphorylation analysis of immunoreceptor tyrosine inhibitory motif signaling associated phosphatases SHP-1 and SHP-2. Phosphoprotein intensity was set in relation to intensity of unphosphorylated protein shown in panels H and J. Representative western blots are shown in panels G and I. All graphs display median ± IQR.
Sepsis platelets show impaired (hem)ITAM signaling. Washed platelets (500 000/µL) of healthy controls (HC) and patients with sepsis (S) were stimulated with CRP-XL (C) and rhodocytin (R). Lysis was done at the following time points: C: 5 minutes; R: 10 minutes. I: admission day; II: day 5 to 7; and III: day of ICU discharge. (A-C) Western blot analysis of platelet lysates. Staining was performed with anti-phospho-tyrosine antibody 4G10. Band intensity (72 kDa) was set in relation to signal intensity of Syk or GAPDH in AU. Representative blots are shown in panels A (time point I) and B (time point III). (D-F) Phosphorylation of signaling peptides Syk and Lat was investigated by immunoblot. One representative blot is shown in panel D. (G-J) Phosphorylation analysis of immunoreceptor tyrosine inhibitory motif signaling associated phosphatases SHP-1 and SHP-2. Phosphoprotein intensity was set in relation to intensity of unphosphorylated protein shown in panels H and J. Representative western blots are shown in panels G and I. All graphs display median ± IQR.
Members of the immunoreceptor tyrosine inhibitory motif protein family are major negative regulators of ITAM-receptor signaling by Src-dependent phosphorylation of protein tyrosine phosphatases SHP-1 and SHP-2, leading to subsequent dephosphorylation of many activated signaling molecules.24,25
The phosphorylation levels of SHP-1 or SHP-2 in resting platelets of sepsis patients were unaltered as compared with controls. Both proteins became phosphorylated upon CRP-XL or rhodocytin stimulation in platelets from healthy donors. In contrast, platelets from patients with sepsis failed to induce SHP-1 or SHP-2 tyrosine phosphorylation (Figure 4G-J). This observation indicates that a constitutive basal SHP-1/2 activity is unlikely to be causative for the activation defect in sepsis platelets and suggests that the sepsis-mediated defect is located upstream in the ITAM-receptor cascades because of the dysfunctional feedback mechanism.
Taken together, this set of experiments strongly implies that the GPVI signaling cascade (including Syk and LAT) is massively impaired, albeit not completely abrogated.
CRP-XL dose escalation restored activation and aggregation of platelets in sepsis
We next asked whether the signaling defect in platelets from patients with sepsis could be overcome by dose escalation.26 Indeed, when a 10-fold excess concentration of CRP-XL was used in aggregometry (and 100-fold in flow cytometry), the response was normalized to control values, as shown by normal CD62P exposure and integrin αIIbβ3 activation. Moreover, the maximal aggregation increased up to reference levels (Figure 5A-D), concomitantly with increased tyrosine phosphorylation levels, detected by immunoblotting (Figure 5E-F). Taken together, these data support our finding that during sepsis the signaling cascades in platelets are markedly attenuated, but not completely abrogated.

CRP-XL dose escalation leads to increased aggregation and activation of platelets in sepsis. Platelet reactivity of healthy controls (HC) and patients with sepsis (S) was depicted upon stimulation with CRP-XL standard dose (C) (0.1 µg/mL in aggregometry; 0.01 µg/mL in flow cytometry) or CRP-XL high dose (CHi) (1 µg/mL) (A-B) Platelet activation due to integrin activation indicated through PAC-1 binding (A) or P-selectin exposure (B) was assessed in whole blood by flow cytometry. (C-D) Light transmission aggregometry was performed using washed platelets (500 000/µL). Samples were measured for 5 minutes. Representative curves are shown in panel C. (E-F) Platelets were lysed after 5 minutes of CRP-XL stimulation or resting conditions. Staining was performed with phosphotyrosine antibody 4G10. Band intensity (72 kDa) was set in relation to a housekeeping protein in AU. Representative blots are shown in panel E (time point I). All graphs show median ± IQR.
CRP-XL dose escalation leads to increased aggregation and activation of platelets in sepsis. Platelet reactivity of healthy controls (HC) and patients with sepsis (S) was depicted upon stimulation with CRP-XL standard dose (C) (0.1 µg/mL in aggregometry; 0.01 µg/mL in flow cytometry) or CRP-XL high dose (CHi) (1 µg/mL) (A-B) Platelet activation due to integrin activation indicated through PAC-1 binding (A) or P-selectin exposure (B) was assessed in whole blood by flow cytometry. (C-D) Light transmission aggregometry was performed using washed platelets (500 000/µL). Samples were measured for 5 minutes. Representative curves are shown in panel C. (E-F) Platelets were lysed after 5 minutes of CRP-XL stimulation or resting conditions. Staining was performed with phosphotyrosine antibody 4G10. Band intensity (72 kDa) was set in relation to a housekeeping protein in AU. Representative blots are shown in panel E (time point I). All graphs show median ± IQR.
Platelet hyporeactivity is not inducible through bacteria or plasmatic factors
The hitherto assessed findings do not allow differentiation whether in septic patients hyporeactive platelets are produced de novo, based on altered megakaryopoiesis, or platelet dysfunction is induced in circulating platelets.
In 14 of 15 patients, at least 1 bacterial strain was identified from distinct material as indicated (see supplemental Table 2). As several strains have been reported to have a direct or indirect effect on platelet function,27,28 we next analyzed whether the GPVI signaling defect was affected by isolated bacteria. We expanded and purified 1 gram-positive (Enterococcus faecalis ID-S29) and 2 gram-negative strains (Escherichia coli ID-S28 and K pneumoniae ID-S23), which were cultured under either colonial or planktonic growth conditions. Platelets were isolated from 3 healthy controls and coincubated with bacteria in a serial dilution of 0.1 to 0.001 OD600. As a positive control, we used a Porphyromonas gingivialis strain, which was recently described to directly induce platelet aggregation.29 Upon coincubation with the highest concentration (OD600 = 0.1) of P gingivalis, platelets were readily activated, as shown by CD62P expression.
Surprisingly, besides P gingivalis, only incubation with E faecalis led to a discrete platelet activation (Figure 6A-D). K pneumonia or E coli did not induce CD62P exposure. After bacterial coincubation, platelets of 3 healthy controls demonstrated an unaltered response to CRP-XL stimulation.

Platelet hyporeactivity is not inducible through bacteria or plasmatic factors but can be triggered by septic whole blood. P-selectin exposure and integrin activation are shown under resting conditions (−) and upon activation with 0.01 µg/mL CRP-XL. (A-D) Whole blood of healthy donors was mixed with isolated patient-borne bacterial strains adjusted to defined optical density at 600 nm (OD600) or with modified HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid)-buffered Tyrode solution (H). CD62P expression was analyzed on resting platelets (−) after 15 minutes. After incubation for 60 minutes, platelets were stimulated with CRP-XL. (E) Platelets were incubated for 60 minutes in PPP of patients with sepsis (S) or healthy controls (HC) (n = 8). (F) Platelets were incubated for 15 minutes in whole blood of patients with sepsis (S) or healthy controls (HC) (n = 7). (A-D) The depicted data represent the mean. (E-F) The data represent mean ± standard error of the mean. Differences were analyzed using Wilcoxon matched-pairs signed rank test. *P < .05.
Platelet hyporeactivity is not inducible through bacteria or plasmatic factors but can be triggered by septic whole blood. P-selectin exposure and integrin activation are shown under resting conditions (−) and upon activation with 0.01 µg/mL CRP-XL. (A-D) Whole blood of healthy donors was mixed with isolated patient-borne bacterial strains adjusted to defined optical density at 600 nm (OD600) or with modified HEPES (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid)-buffered Tyrode solution (H). CD62P expression was analyzed on resting platelets (−) after 15 minutes. After incubation for 60 minutes, platelets were stimulated with CRP-XL. (E) Platelets were incubated for 60 minutes in PPP of patients with sepsis (S) or healthy controls (HC) (n = 8). (F) Platelets were incubated for 15 minutes in whole blood of patients with sepsis (S) or healthy controls (HC) (n = 7). (A-D) The depicted data represent the mean. (E-F) The data represent mean ± standard error of the mean. Differences were analyzed using Wilcoxon matched-pairs signed rank test. *P < .05.
Taken together, these data suggest that the defective GPVI/ITAM signaling was not directly caused by platelet-bacterial interactions.
As all patients with sepsis received antibiotic therapy, we also evaluated whether antimicrobial drugs could reduce GPVI/CLEC-2 reactivity. We selected the commonly used antibiotics piperacillin/tazobactam, ciprofloxacin, metronidazole, and vancomycin, which differ in mode of action and do not require first-pass activation (supplemental Table 2) for incubation with whole blood of healthy donors at therapeutic peak plasma levels. None of these drugs had an impact on GPVI-mediated platelet reactivity (supplemental Figure 5), making it unlikely that any of the used antibiotic drugs could account for the observed platelet dysfunction.
To investigate whether plasmatic or cellular factors of patients with sepsis impair platelet function, we performed incubation experiments using platelet poor plasma (PPP) and whole blood of sepsis patients. Incubation of resting platelets from healthy donors in plasma of patients with sepsis did not lead to elevated CD62P expression or PAC-1 binding. Moreover, preincubation in sepsis plasma did not cause a diminished response to CRP-XL or TRAP-6 agonists (Figure 6E; supplemental Figure 6A-B). Freshly isolated PPP or cryopreserved PPP led to comparable results, as confirmed by flow cytometry or aggregometry (supplemental Figure 6C). Our data thus imply that platelet hyporeactivity during sepsis is not primarily induced by plasmatic factors. In contrast, preincubation of antibody-marked healthy donor platelets in whole blood of septic patients clearly diminished the response toward CRP-XL, suggesting that the abrogated GPVI signaling is partially (co-)mediated by cellular components (Figure 6F).
Discussion
In this pilot cohort study, we performed a comprehensive platelet function analysis in 15 consecutive patients suffering from sepsis or septic shock. Despite the small cohort size, with limited underlying infection types and sites, we have found a surprisingly uniform and pronounced hyporeactivity of platelets to multiple agonists. As a result of the longitudinal assessment of platelet function in this prospective study, we hypothesize that acquired platelet function to various agonists, but especially to GPVI, might be an early event during sepsis onset and disease progression.
Although our data demonstrate that platelet hyporeactivity was most pronounced upon stimulation with the GPVI-agonist CRP-XL, we cannot rule out that other receptors (like CD42b) might modulate the signal strength.23 GPVI and its downstream signaling cascade of the SFK-Syk-LAT-PLCγ2 pathway is highly relevant for the maintenance of vascular integrity7 and plays a beneficial role for host defense.9 Its dysfunction might contribute to inflammatory immune responses30 and progression into capillary leakage syndrome. Using rhodocytin, a comparable platelet signal transduction defect was evident, implying that also the signaling cascade downstream of CLEC-2 is affected. It remains unclear whether downregulation of the common ITAM-signaling pathway occurs to prevent overwhelming activation because of the sudden increase in activating ligands (like histones, fibrin, or collagen) exposed in response to vessel damage or the inflammatory condition.9 By this means, an unwanted consumption of peripheral platelets could be counteracted.
Results from mouse studies provide evidence that ectopic expression of podoplanin, the only yet identified physiological ligand for CLEC-2, can be found on leukocyte subsets in response to a septic trigger. CLEC-2–leukocyte interactions are involved in platelet dysfunction during sepsis10 and can trigger the release of complement inhibitors from platelet granules.31 In our study, we could not detect any significant increase in podoplanin expression on CD14+/CD45+ leukocytes (supplemental Figure 8A). Nonetheless, we found an increased ratio of CD41a+/CD45+ complexes as a marker for platelet-leukocyte aggregates (supplemental Figure 8B). This is in line with previous studies15 and suggests that platelets might become activated because of direct contact with leukocytes. The role of (hem)ITAM signaling in this matter, however, remains unclear.
A growing body of evidence supports the importance of platelets for the regulation of inflammation and host defense. Claushuis and colleagues have shown an impaired immune response of GPVI−/− mice in a K pneumoniae inhalation mouse model, whereas CLEC-2 depletion did not affect responses during induced pneumonia.9 In contrast, Rayes and coworkers could not find a role for GPVI, but for CLEC-2 in systemic inflammation and organ damage, using 2 mouse models of sepsis–intraperitoneal lipopolysaccharide and cecal ligation and puncture.10 Although mouse models and bacterial strains differ, there is strong evidence that the signaling cascade shared by ITAM and hemITAM receptors is involved in septic inflammation.11 Furthermore, platelets store antimicrobial proteins and complement inhibitors known to be bactericidic to some species, including Staphylococcus aureus, Bacillus subtilis, or E coli.30,31 Because α-granule release is most efficient when in close contact to bacteria, a microbe-dependent platelet activation should occur to some extent. We tested whether the impaired platelet function could be explained by a direct platelet-bacteria interaction. Although stimulation with P gingivalis could activate platelets,29 also in our experimental setup, none of the distinct patient-borne bacterial strains could induce hyporeactivity in platelets of healthy donors or directly activate healthy platelets (in their own blood or plasma), implying that the bacteria themselves are not the key trigger for the hyporeactivity. This finding is independently supported by the fact that several gram-positive and -negative strains were used, making a common bacteria-mediated mechanism for downregulation of GPVI signaling unlikely. Furthermore, we did not find evidence that the antimicrobial drugs used in treatment of our patients were accountable for the platelet dysfunction, although we are fully aware that this is difficult to assess due to the small cohort size. Although preincubation of healthy platelets in plasma of patients with sepsis did not impair their reactivity, preincubation in whole blood significantly reduced their response to CRP-XL, albeit not to the same extent as in sepsis patients. This finding implies that platelet hyporeactivity is partially inducible in healthy platelets and further modulated by cellular interactions. Investigating the full mechanism will require further studies.
We found GPVI receptor ectodomain shedding could account, at least in part, for the observed platelet hyporeactivity,32-34 which has recently been shown in trauma patients.35 Shedding of surface receptors is a common mechanism involved in downregulating of various signal-transduction pathways.36 Although elevated plasma levels of soluble GPVI are discussed as a sepsis biomarker,37 in our cohort, we did not find a correlation between GPVI expression, sGPVI plasma levels, and the observed GPVI signaling deficiency. These findings do not support the hypothesis of GPVI receptor shedding from the platelet surface.
A recent report provides evidence that GPVI signaling is partially inactive in newly formed platelets but gets (re)activated when platelets are released into the circulation.38 In our study, we found elevated TPO plasma levels, an increased mean platelet volume, as well as an increased fraction of TO-positive platelets as a marker for stimulated thrombopoiesis. However, our results by flow cytometry indicate that from sepsis onset on, the response to GPVI agonists (especially integrin activation) was uniformly reduced in the entire platelet population. This observation points to a mechanism that affects all circulating platelets rather than a gradual shift toward the predominance of a platelet population with hyporeactivity or the selective loss of a platelet subfraction. Middleton and colleagues have recently demonstrated that human and mouse platelets change their gene expression profile under septic conditions.39 As the described GPVI defect in our patients was detectable already hours after sepsis onset, we consider that altered biogenesis is most likely not the major source of hyporeactive platelets, at least in our cohort.
Interestingly, the value of maximal aggregation upon CRP-XL stimulation increased as the patients recovered. This tendency was not found when we studied this agonist by whole blood flow cytometry. This discrepancy could be explained by the fact that a few functional integrin molecules are sufficient to induce platelet aggregation.40 Regarding the new outstanding data on sepsis phenotypes, most of our study patients show a pattern that is typical for the δ-phenotype according to the laboratory pattern, the high mortality, and the high prevalence of septic shock.41
Our study has several limitations: (1) Our patient number is rather small. The main interest was to provide a comprehensive and complementary set of platelet function tests and agonists rather than performing standard stimulations like CD62P with few agonists. A second focus was to follow these broad platelet function results during disease progression, not knowing whether patients would aggravate or recover. We thus decided to design a hypothesis-forming pilot study with an extensive array of tests over time. Further studies with larger cohorts are required to corroborate our selective GPVI signaling defect. (2) All our patients are of white descent, but we did not consider GPVI variants. Two frequent haplotypes (GP6a and GP6b) have been reported, resulting in protein variants GPVIa (a/a), GPVIb (a/b), and GPVIc (b/b). GPVIc expression results in a slightly diminished GPVI surface expression and reduced reactivity.42-44 In a recent pediatric sepsis cohort study, patients with GPVIb showed increased platelet microparticle formation and an overall worse outcome compared with patients with GPVIa.45 We could determine that 5 of 7 of our patients (where DNA was available) had GPVIa and 2 of 7 had GPVIb variants (supplemental Table 1), without any obvious difference in reactivity (data not shown), suggesting that the distinct GP6 haplotypes are unlikely to explain why all patients in our cohort show a virtually identical defect in (hem)ITAM signaling. (3) Finally, our study was not powered to control for survival as a major end point. The clear separation of survivors vs nonsurvivors with restored GPVI signaling (supplemental Figure 9), however, is an observation worth following in future studies.
In summary, our study provides several lines of evidence that different platelet signaling pathways are severely dysregulated during sepsis and that especially the defective response to GPVI agonists could be exploited as an early biomarker for the diagnosis of sepsis.
For original data, please contact the corresponding author at harald.schulze@uni-wuerzburg.de.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Goetz Ulrich Grigoleit for his support as consulting physician, as well as Heike Claus and Ulrich Vogel for their support with the bacterial experiments. Thanks to David Stegner and Andreas Starke for establishing the sGPVI-ELISA, to Attila Braun for comments on the manuscript, and to Irina Pleines for language editing and critical comments on the manuscript. Eva Klopocki helped with the GP6 haplotype analysis.
This work was funded, in part, by the Deutsche Forschungsgemeinschaft (German Research Foundation), Projektnummer 374031971-TRR 240 (H.S., B.N.). L.J.W., G.M., D.W., and H.S. have filed a patent (EP19190817.7) related to this study.
Authorship
Contribution: L.J.W. designed and performed experiments, analyzed data, and wrote the manuscript; G.M. designed experiments, analyzed data, and wrote the manuscript; A.P. designed and performed experiments; N.W. performed experiments; N.N., M.K., and D.W. designed the study and cared for study patients; M.W. analyzed data; B.N. provided reagents and analyzed data; T.-T.L. designed experiments and analyzed data; H.S. designed the concept and experiments, analyzed data, and wrote the manuscript; and all authors critically revised and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Harald Schulze, Institute for Experimental Biomedicine, University Hospital Würzburg, Josef-Schneider-Strasse 2/D15, 97080 Würzburg, Germany; e-mail: harald.schulze@uni-wuerzburg.de.








