In this issue of Blood, present an immunophenotypic and genomic characterization of samples from 114 patients with suspected chronic lymphoproliferative disorders of natural killer cells (CLPD-NK). They report a high incidence of TET2 mutations, including the detection of these as a postulated precursor cell lesion in the myeloid compartment.1 As a novel biologic and diagnostic hallmark of CLPD-NK, this finding provides a clinically useful marker for distinguishing CLPD-NK from reactive NK-cell expansions as well as for the discrimination of defined CLPD-NK subsets. The bilinear presence of TET2 variants substantiates existing epidemiological links between CLPD-NK and myeloid disorders.
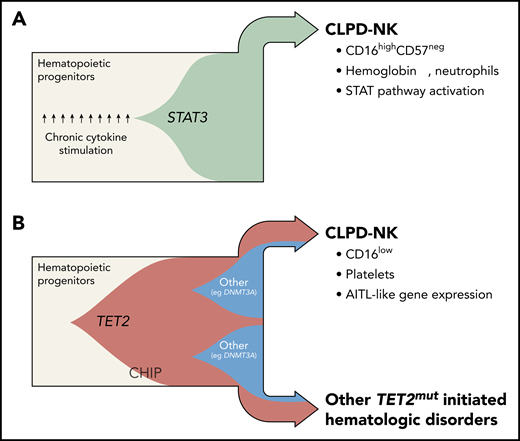
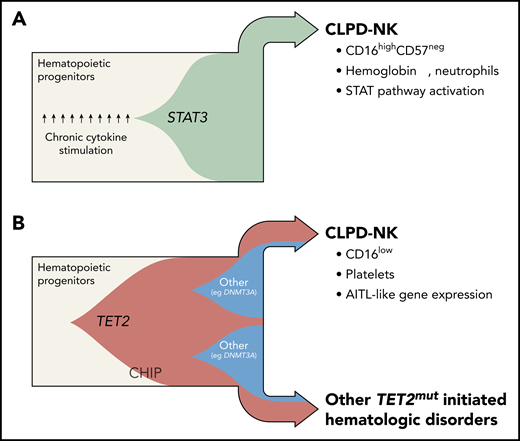
The discovery of genomic TET2 variants in CLPD-NK has redefined the understanding of the clonal evolution in 2 major subsets of the disease. Fish plots capture the postulated clonal hierarchy of STAT3-mutated (A) and TET2-mutated (B) CLPD-NK. Until recently, the mechanistic model of CLPD-NK had been centered on pronounced cytokine exposure of the precursor cell and/or its gain-of-function STAT3 SH2 domain mutations, both leading to constitutive STAT activation (beige = myeloid/lymphoid precursor cells; green = STAT3-mutated clone). Pastoret et al provide evidence of highly recurrent loss-of-function TET2 mutations in CLPD-NK (34%) and in the myeloid compartment of affected cases. Such TET2 lesions are likely to have their origin in common progenitor cells. Generally, they are frequent nonconsequential constituents of a CHIP and are also likely under the right conditions to give rise to myeloid and NK/T-cell neoplasms (beige = myeloid/lymphoid precursor cells; red = TET2-mutated clone; blue = DNMT3A-mutated clone). TET2-mutated cases are obviously distinct, histogenetically and clinically. In fact, TET2-mutated cases presented with lower platelet counts, a CD16low phenotype, an “AITL-like” transcriptome, and more frequently other malignant hematologic diseases, whereas the almost mutually exclusive STAT3-mutated cases (27%) were characterized by anemia, lower neutrophil counts, a cytotoxic CD16highCD57neg pattern, and a gene expression signature that resembles STAT pathway activation. Professional illustration by Somersault18:24.
The discovery of genomic TET2 variants in CLPD-NK has redefined the understanding of the clonal evolution in 2 major subsets of the disease. Fish plots capture the postulated clonal hierarchy of STAT3-mutated (A) and TET2-mutated (B) CLPD-NK. Until recently, the mechanistic model of CLPD-NK had been centered on pronounced cytokine exposure of the precursor cell and/or its gain-of-function STAT3 SH2 domain mutations, both leading to constitutive STAT activation (beige = myeloid/lymphoid precursor cells; green = STAT3-mutated clone). Pastoret et al provide evidence of highly recurrent loss-of-function TET2 mutations in CLPD-NK (34%) and in the myeloid compartment of affected cases. Such TET2 lesions are likely to have their origin in common progenitor cells. Generally, they are frequent nonconsequential constituents of a CHIP and are also likely under the right conditions to give rise to myeloid and NK/T-cell neoplasms (beige = myeloid/lymphoid precursor cells; red = TET2-mutated clone; blue = DNMT3A-mutated clone). TET2-mutated cases are obviously distinct, histogenetically and clinically. In fact, TET2-mutated cases presented with lower platelet counts, a CD16low phenotype, an “AITL-like” transcriptome, and more frequently other malignant hematologic diseases, whereas the almost mutually exclusive STAT3-mutated cases (27%) were characterized by anemia, lower neutrophil counts, a cytotoxic CD16highCD57neg pattern, and a gene expression signature that resembles STAT pathway activation. Professional illustration by Somersault18:24.
In the current World Health Organization classification of hematologic neoplasms,2 CLPD-NK represents a distinct entity within the category of large granular lymphocyte leukemias (LGL). Patients with CLPD-NK typically present with expansions of mature NK cells, which display an LGL morphology and a restricted killer immunoglobulin-like receptor (KIR) pattern.
The rarity and the NK-cell nature of CLPD-NK impose major diagnostic obstacles. The lack of antigen-receptor gene rearrangements, normally a reliable readout for clonality in T-lineage neoplasms, such as T-LGL, requires the use of KIR phenotyping for diagnosis. However, this is not a well-established technique in routine diagnostic laboratories. Until this report by Pastoret et al, somatic STAT3 mutations have been considered the most reliable genomic marker in the diagnostic workup of CLPD-NK. However, these lesions were only found in ≈30% of cases and often at low allelic fractions.3 Moreover, such mutations, formerly thought to provide evidence for the clonal nature of a given process, have been increasingly detected in chronic inflammatory conditions, for example, in rheumatoid arthritis (RA), with recurrent STAT3 lesions found in the autoreactive cytotoxic T cells.4
Pastoret et al performed targeted high-throughput sequencing of samples from a cohort of patients with NK-cell expansions. They retrospectively classified these as 46 cases of CLPD-NK and 68 cases of reactive NK-cell expansions. Loss-of-function TET2 mutations were detected in 34% of CLPD-NK. A TET2 mutation was previously described by Raess et al5 in a single case of NK-LGL and by Gasparini et al6 in 2 cases NK-LGL. The present paper also confirmed the recurrence of activating STAT3 variants by detecting them in 27% of CLPD-NK. Interestingly, TET2 and STAT3 lesions were almost mutually exclusive, as only 1 sample was identified as doubly mutated. In addition, rarer mutations were detected in TNFAIP3 (10%), DNMT3A (7%), NRAS or KRAS (7%), and STAT5B (5%).
The authors integrated these findings to generate an algorithm to better classify NK-cell proliferations. Their new NK clonality score is based on the NK-cell blood count as well as on immunophenotypic (KIR restriction, elevated expression of CD94 and NKG2A) and genomic data (at least 1 mutation in TET2, STAT3, STAT5B, and/or TNFAIP3). This score performed robustly in discriminating between patients with CLPD-NK and those with reactive NK proliferations in an independent validation cohort.
It is intriguing that the 2 major CLPD-NK subsets, namely those of mutated TET2 and STAT3, differed markedly in phenotypic and clinical features.1 STAT3-mutated CLPD-NK predominantly presented with a cytotoxic CD16highCD57neg profile, lower neutrophil counts, decreased hemoglobin levels, and a gene expression signature of STAT pathway activation. Conversely, CLPD-NK with TET2 mutations exhibited a CD16low phenotype, presented with lower platelet counts, and showed an “angioimmunoblastic T-cell lymphoma (AITL)-like” transcriptome.
Of great interest in the field of leukemogenesis is the role of TET2 lesions as ancestral events. Pastoret et al demonstrated that TET2 mutations coexist in the myeloid compartment in 4 of 6 CLPD-NK. No TET2 mutations were detected in the T-cell lineage. Moreover, the incidence of cooccurring hematologic malignancies was increased in the TET2-mutated cohort; in fact, at least 1 concomitant hematologic malignancy was detected in 38% of the patients with TET2-mutated CLPD-NK, whereas only 7% of the TET2 wild-type cohort showed a second malignant disorder. One patient developed AITL 2 years prior to the diagnosis of CLPD-NK, and both neoplasms shared the same TET2 mutation. Together, this adds important data to the reported associations of myeloid neoplasms7 or clonal hematopoiesis of indeterminate potential (CHIP)8 with AITL, believed to be due to a common TET2/DNMT3A molecular trunk with subsequently divergent context-dependent oncogenic branches (see figure). Such a conceptual framework also provides for alternative paths for NK-cell ontogeny, which might not be exclusively lymphoid, but also of myeloid origin.9
It is also important to highlight that the NK-cell proliferations that were categorized as ”reactive” showed DNMT3A mutations in 13% and TET2 mutations in 7% of cases.1 Fittingly, these 2 genomic variants were identified as the most frequent in the CHIP that accompany active RA.10
This paper indicates that there are 2 distinct pathogenetic routes for CLPD-NK: (1) up to this point, the mechanistic model of CLPD-NK had focused on constitutive STAT activation, either based on pronounced cytokine exposure of the precursor or via activating STAT3 mutations; and (2) although confirming STAT3/5B variants in approximately one-third of cases (32%), Pastoret et al provide the first evidence of loss-of-function variants of TET2 in another third (34%) of CLPD-NK. They illustrate the cooccurrence of mutated TET2 in the myeloid lineage and postulate a TET2-based ancestral trajectory that is expanded and refined in the figure.
Collectively, the landmark paper by Pastoret et al is an important contribution to the fields of LGL and of general leukemogenesis. It establishes TET2 mutations as a diagnostic marker in CLPD-NK, including its use in a new clonality score or as an informative lesion to define a clinicopathological subset of CLPD-NK that is set apart from cases with an underlying STAT signature. Pastoret et al also have set the stage for further exploration (eg, single-cell resolved approaches to sequential samples) to refine our concepts of lesion-based clonal relationships in perturbed hematopoiesis and its translational implications. Related questions include the leukemogenic factors that cooperate with mutated TET2 to increase its penetrance and provision of context-specificity (ie, lineage commitment), such as particular comutations or permissive milieus of protracted inflammation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.