Abstract
Platelets have been hypothesized to promote certain neoplastic malignancies; however, antiplatelet drugs are still not part of routine pharmacological cancer prevention and treatment protocols. Paracrine interactions between platelets and cancer cells have been implicated in potentiating the dissemination, survival within the circulation, and extravasation of cancer cells at distant sites of metastasis. Signals from platelets have also been suggested to confer epigenetic alterations, including upregulating oncoproteins in circulating tumor cells, and secretion of potent growth factors may play roles in promoting mitogenesis, angiogenesis, and metastatic outgrowth. Thrombocytosis remains a marker of poor prognosis in patients with solid tumors. Experimental data suggest that lowering of platelet count may reduce tumor growth and metastasis. On the basis of the mechanisms by which platelets could contribute to cancer growth and metastasis, it is conceivable that drugs reducing platelet count or platelet activation might attenuate cancer progression and improve outcomes. We will review select pharmacological approaches that inhibit platelets and may affect cancer development and propagation. We begin by presenting an overview of clinical cancer prevention and outcome studies with low-dose aspirin. We then review current nonclinical development of drugs targeted to platelet binding, activation, and count as potential mitigating agents in cancer.
Introduction
Platelets are anucleate cells released into the bloodstream from proplatelet extensions of megakaryocytes. Part hemostatic sentinel, part mitogenic agent, these centaurs of the circulation play essential roles in maintaining vascular integrity and promoting wound healing. Paradoxically, these physiological attributes may play a deleterious role in empowering the growth and dissemination of tumor cells.1-5 Platelets have been implicated in enabling successful metastasis and worsening the prognosis of patients with cancer by guarding tumor cells from immune elimination and promoting arrest and extravasation of tumor cells (Figure 1).3,5-11 In a symbiotic manner, tumor-derived bioactive molecules have been shown to prompt an increase in platelet activation and production.12-18 Recent epidemiological and computational studies have shown an association of temporally derived platelet count features with prognosis for patients with lung, prostate, and colon cancers.19-23 Therefore, strategies aimed at reducing the activation, activity, or number of platelets have been explored as anticancer therapies. This review provides an overview of the history and use of antiplatelet strategies to combat or manage cancer and metastasis.
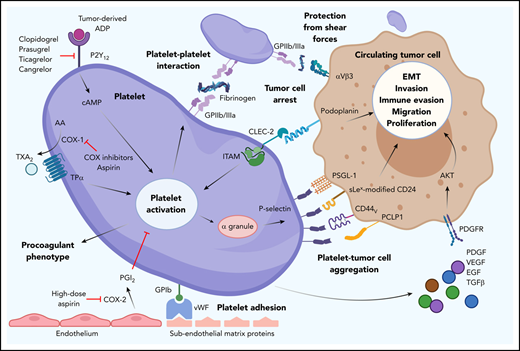
![Overview of platelet and tumor cell interaction. Platelet activation can occur by many routes, initiated here by tumor-derived adenosine diphosphate (ADP) interaction with platelet P2Y12, which can be blocked by thienopyridines. Endothelial damage exposes subendothelial matrix proteins, allowing von Willebrand factor (vWF) to bind and tether to platelet GPIb. Cyclooxygenase (COX) inhibitors prevent platelet-expressed COX-1 production of TXA2. Aspirin also inhibits COX-2–mediated endothelial prostacyclin (PGI2) production and platelet adhesion. Upregulation of GPIIb/IIIa allows platelet-platelet and platelet–tumor cell aggregation. Upregulation of P-selectin from platelet α granules interacts with many ligands on tumor cells (eg, PSGL-1, sialyl-Lewisx–modified CD24 [sLex-modified CD24], CD44 variant [CD44v], PCLP1). Platelet release of platelet-derived growth factors (eg, platelet-derived growth factor [PDGF], vascular endothelial growth factor [VEGF], epithelial growth factor [EGF], transforming growth factor β [TGFβ], and cytokines) promotes tumor cell immune evasion, migration, epithelial-mesenchymal transition (EMT), invasion, and proliferation. AA, arachidonic acid; cAMP, cyclic adenosine monophosphate; CLEC-2, C-type lectin-like receptor 2; PCLP1, podocalyxin-like protein 1; PDGFR, PDGF receptor; PGI2, prostacyclin; PSGL-1, P-selectin glycoprotein ligand-1; TPα, thromboxane receptor α; TXA2, thromboxane A2.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/23/10.1182_blood.2019003977/5/m_bloodbld2019003977cf1.png?Expires=1769606432&Signature=Cfc9KRlHSBfp0lM-LXGVmcRcvbSTzN7f8aNLsOw9u4bBWykKdcWQL4e2rBtZKlHQZOPXwwVg84T4c0wqD~8Fo15NHpLuPVm9HNoCvYBKwwxfSz1WYC-hKmXBC4GtAVG~4enFEFjqkc-wSCFmwqQ0gtqFkGRNSy3oyxSjzOmifeSOQeELzIG1kYcLNXRWd~NNlgKuX3rwWx3cltok21TLCac2B43WcCy2RjC3d3nEHTlcipQEL9kPo1mKL-IpWZfzrn-obqKRfHUIySKjMIY4KJQIwjPFzzVeOlgeDmnXeHlCQw0u9OEY8wUiNFm80---NWMO1fEADoDAGIJolhaXYw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Overview of platelet and tumor cell interaction. Platelet activation can occur by many routes, initiated here by tumor-derived adenosine diphosphate (ADP) interaction with platelet P2Y12, which can be blocked by thienopyridines. Endothelial damage exposes subendothelial matrix proteins, allowing von Willebrand factor (vWF) to bind and tether to platelet GPIb. Cyclooxygenase (COX) inhibitors prevent platelet-expressed COX-1 production of TXA2. Aspirin also inhibits COX-2–mediated endothelial prostacyclin (PGI2) production and platelet adhesion. Upregulation of GPIIb/IIIa allows platelet-platelet and platelet–tumor cell aggregation. Upregulation of P-selectin from platelet α granules interacts with many ligands on tumor cells (eg, PSGL-1, sialyl-Lewisx–modified CD24 [sLex-modified CD24], CD44 variant [CD44v], PCLP1). Platelet release of platelet-derived growth factors (eg, platelet-derived growth factor [PDGF], vascular endothelial growth factor [VEGF], epithelial growth factor [EGF], transforming growth factor β [TGFβ], and cytokines) promotes tumor cell immune evasion, migration, epithelial-mesenchymal transition (EMT), invasion, and proliferation. AA, arachidonic acid; cAMP, cyclic adenosine monophosphate; CLEC-2, C-type lectin-like receptor 2; PCLP1, podocalyxin-like protein 1; PDGFR, PDGF receptor; PGI2, prostacyclin; PSGL-1, P-selectin glycoprotein ligand-1; TPα, thromboxane receptor α; TXA2, thromboxane A2.
Overview of platelet and tumor cell interaction. Platelet activation can occur by many routes, initiated here by tumor-derived adenosine diphosphate (ADP) interaction with platelet P2Y12, which can be blocked by thienopyridines. Endothelial damage exposes subendothelial matrix proteins, allowing von Willebrand factor (vWF) to bind and tether to platelet GPIb. Cyclooxygenase (COX) inhibitors prevent platelet-expressed COX-1 production of TXA2. Aspirin also inhibits COX-2–mediated endothelial prostacyclin (PGI2) production and platelet adhesion. Upregulation of GPIIb/IIIa allows platelet-platelet and platelet–tumor cell aggregation. Upregulation of P-selectin from platelet α granules interacts with many ligands on tumor cells (eg, PSGL-1, sialyl-Lewisx–modified CD24 [sLex-modified CD24], CD44 variant [CD44v], PCLP1). Platelet release of platelet-derived growth factors (eg, platelet-derived growth factor [PDGF], vascular endothelial growth factor [VEGF], epithelial growth factor [EGF], transforming growth factor β [TGFβ], and cytokines) promotes tumor cell immune evasion, migration, epithelial-mesenchymal transition (EMT), invasion, and proliferation. AA, arachidonic acid; cAMP, cyclic adenosine monophosphate; CLEC-2, C-type lectin-like receptor 2; PCLP1, podocalyxin-like protein 1; PDGFR, PDGF receptor; PGI2, prostacyclin; PSGL-1, P-selectin glycoprotein ligand-1; TPα, thromboxane receptor α; TXA2, thromboxane A2.
Aspirin use in cancer therapy
The use of aspirin in cancer therapy was built upon the observation that nonsteroidal anti-inflammatory drugs (NSAIDs) suppressed chemical-induced carcinogenesis.24,25 Although the mechanistic pathways were unclear, inhibition of COX, the primary mechanism of NSAIDs, was presumed to play a role. Because aspirin is an irreversible inhibitor of the 2 isoforms of COX, epidemiological trials soon began looking for a similar benefit.26-28 Thus began the inquiry into whether an old drug could provide a new benefit; the ensuing clinical trials are summarized in Table 1.
Ancient Egyptians used extracts of the willow tree, which contains salicin, an aspirin prodrug, to relieve body aches; aspirin as we know it today was first introduced in 1897 when Felix Hoffmann added an acetyl group to salicylic acid to produce acetylsalicylate powder.35 Aspirin, like other NSAIDs, blocks the conversion of arachidonic acid to prostanoids by inhibiting COX enzymes, an inhibitory effect that is dependent on the effective dose administered.36 Low-dose aspirin (75-100 mg per day; 15-20 μM of salicylic acid in plasma) irreversibly inhibits COX-1, thereby reducing the ability of platelets to produce and secrete the secondary platelet agonist thromboxane A2. At higher doses, aspirin also wields anti-inflammatory effects via inhibition of both COX-1 and COX-2. Leukocytes, endothelial cells, mucosal cells, and vascular smooth muscle cells express this second isoform of COX. Selective targeting of COX-2 suppresses the prostaglandins, particularly prostacyclin, at sites of vascular inflammation. In cancer, the possible mechanisms by which aspirin may provide benefit range from a direct inhibitory effect on cancer cells themselves to antiplatelet effects, including reducing platelet–tumor cell interactions or reducing platelet secretion of proangiogenic and growth factors, cytokines, and chemokines. Moreover, in vitro studies have shown that the ability of platelets to potentiate upregulation of oncoproteins, including c-MYC, in cancer cells is sensitive to aspirin, providing yet another anticancer effect resulting from the effect of aspirin on platelets.37 However, the potential anticancer benefits of aspirin may likewise be due to direct effects on blood and vascular cells, as well as on cancer cells themselves within a primary or secondary tumor or during circulation in the vasculature.18,38-42 For instance, malignant tumors within the proinflammatory and antiapoptotic tumor microenvironment have been shown to aberrantly express COX-1 and COX-2.43,44 Therefore, aspirin may exert an antitumor effect by way of a COX-related inhibition of inflammation and apoptosis.38 The extent of this effect would likely vary by tumor subtype; for instance, the relative expression COX-1 and COX-2 in ovarian cancer was shown to vary by the histological grade and subtype of the cancer.44 In addition, COX-independent mechanisms have been suggested, including the suppression of signaling by IκB kinase β and extracellular signal-regulated kinase, leading to reduced inflammation and proliferation.45,46 Just as the clinical effects of aspirin on COX-1 vs COX-2 enzymes are dependent on dose and duration, any potential benefit of aspirin in directly reducing the growth of primary tumors, as compared with inhibiting platelets and the seeding or growth of metastatic tumors, likely also depends on factors such as dose, duration, and even the specific form of cancer.37,47-49
Although early retrospective analyses of small trials were equivocal, the Cancer Prevention Study II published in 1991 showed a 40% reduction in colon cancer mortality associated with the regular use of aspirin in a cohort of 662 424 patients.28 Prospective trials were immediately designed, and in 2003, 2 trials published in the New England Journal of Medicine demonstrated clear benefits of low-dose aspirin in secondary prevention of CRC.29,50 Benefits were evident at 1 year, with relative risk reductions of ∼25% for the recurrence of CRC. An accompanying editorial stated that the benefits of aspirin were now proven, at least for CRC.51
However, the question of primary prevention remained open. Despite unclear mechanistic understandings, it was possible that aspirin could suppress metastasis or tumor progression in patients with cancer but not suppress tumorigenesis in patients without cancer.47 Retrospective analyses were conducted once again. This time, large trials designed to study the CVD benefit of aspirin were reexamined for signs of cancer benefit.52-54 However, occurrence of cancer in these trials was low, and the findings of benefit were inconsistent.
While these analyses were ongoing, a recommendation from the US Preventive Services Taskforce (USPSTF) released in 2007 recommended against the routine use of aspirin for any cancer prevention.55 Shortly after the USPSTF recommendation, a large meta-analysis of prospective trials of aspirin for CVD was published, which found a clear benefit of aspirin in reducing both cancer incidence and mortality.56 Unlike the findings for secondary cancer prevention, benefits in primary prevention took years to manifest and were modest. Additional meta-analyses soon followed, expanding the number of trials and strengthening the findings of benefit.57
With these additional data, the USPSTF performed its own analysis. In 2016, it recommended that many adults between the ages of 50 and 69 years would in fact derive cancer benefit from the preventive use of low-dose aspirin, defined as ≤325 mg per day.48 However, the benefit in patients without a history of cancer was small and outweighed by the risk of major bleeding.58 The USPSTF estimated that use of low-dose aspirin in patients between the ages of 50 and 69 years would prevent 15 cases of CRC for every 1000 patients treated for at least 10 years.48 However, aspirin use would also cause 25 major bleeds, of which 2 to 3 would be intracranial hemorrhage.
Clinicians hesitated while larger prospective trials were conducted. In June 2017, the steering committee for the Aspirin in Reducing Events in the Elderly (ASPREE) trial stopped the intervention arm early. The trial was published jointly in 3 reports in 2018.34,59,60 Although the primary outcome was a composite of death and disability, the cancer findings tracked closely with the overall study findings and went against aspirin.34 One month before the publication of ASPREE, the Aspirin to Reduce Risk of Initial Vascular Events (ARRIVE) trial also found an increase in cancer incidence with aspirin.33
The ARRIVE trial enrolled nearly 13 000 patients with a mean age of 64 years. Designed as a cardiovascular trial, ARRIVE did not include cancer as a prespecified end point but did track the incidence of cancer as part of the monitoring of study patients. Patients in ARRIVE were randomly assigned to 100 mg of enteric-coated aspirin or placebo and followed up for an average of 5 years. Differences in cancer incidence were not significant but favored placebo. Prostate cancer occurred in 59 patients (0.94%) receiving aspirin vs 44 (0.70%) receiving placebo, and colon cancer occurred in 14 (0.22%) receiving aspirin and 6 (0.10%) receiving placebo. The authors concluded that the incidence of cancer was too low and the follow-up period of the study was too short to allow meaningful conclusions about the effect of aspirin on the incidence of cancer.33
Published 1 month later, the ASPREE trial was larger than ARRIVE and enrolled an older population, presumably at higher risk for cancer. Among the 19 114 patients randomly assigned to 100 mg of enteric-coated aspirin vs placebo, half were age >74 years.61 Additionally, 56% were women, and 19% had a personal history of cancer.61 The average follow-up time was 4.7 years, a little shorter than that in the ARRIVE trial. Surprisingly, aspirin was associated with an increase in all-cause mortality (HR, 1.14; 95% CI, 1.01-1.29), which was driven largely by an increase in deaths resulting from cancer (HR, 1.31; 95% CI, 1.10-1.56).34 The increased mortality was seen across cancer types, including CRC, melanoma, and breast, brain, and pancreatic cancers. Although the findings were only significant for CRC (HR, 1.77; 95% CI, 1.02-3.06), the trend favoring placebo was found for all cancer types.
Of interest, the increase in cancer-related mortality was not greater among the 19% of patients with a history of cancer.61 Although cancer mortality increased by 31% with aspirin, cancer incidence only increased by 3% (981 participants receiving aspirin vs 952 receiving placebo). Aspirin thus worsened cancer survival substantially more than cancer incidence. It should also be noted that whereas ARRIVE enrolled a population that was >97% white, ASPREE included ∼1500 patients who self-identified as either Black or Hispanic. Among those minority patients, there was no statistical heterogeneity in the primary outcome of cancer mortality.34 The magnitude of effect was lowest among Hispanic patients; however, there were only a total of 12 deaths resulting from cancer in this population.
The cancer findings from ASPREE were surprising and contrasted with multiple previous trials that formed the basis for the 2016 USPSTF recommendations.48 The much smaller increase in cancer incidence compared with cancer mortality and the lack of increased mortality in patients with preexisting cancer suggest that the biological effects of aspirin may differ based on timing of exposure, such as whether it is during tumor initiation and tumorigenesis or during growth or spread of an already established tumor. Because ASPREE had <5 years of follow-up, it is possible that any benefit in reduced tumorigenesis had yet to emerge. Simultaneously, the older age of participants in the ASPREE trial may have led to a higher incidence of early, undiagnosed cancers. If aspirin indeed has any accelerating effect on early cancer growth, perhaps independent of platelet activation and rather as a consequence of direct inhibition of COX in tumor cells, this could offset any beneficial antitumorigenic effects of inhibiting platelet function that had not yet had time to emerge.
The older age of participants enrolled in ASPREE means that the findings are not necessarily incompatible with the 2016 USPSTF report, which recommended consideration of aspirin in patients 50 to 69 years of age.48 Further characterization of the cancers using molecular analysis and death adjudication in ASPREE may lead to a better understanding of whether the effects of aspirin may differ by age or staging of tumor. These questions may be addressed in part by ASPREE-XT,62 a follow-up observational study that is under way and anticipated to be completed in 2024, as well as several ongoing trials of aspirin in patients with cancer.
Clinical evidence supporting the role of aspirin in cancer prevention is greatest for CRC, as in the CAPP2 trial for Lynch syndrome63 ; however, there is mounting evidence in several other cancer types as well. Hepatocellular carcinoma rates were lower among patients with chronic viral hepatitis with low-dose aspirin use.64 Although data supporting the role of aspirin in other cancer types such as melanoma,65 pancreatic cancer,66 and ovarian cancer66,67 have been largely observational, results of several ongoing clinical trials may shed more light on this topic. The ASCOLT ASpirin in Dukes B and C COLorectal cancer (ASCOLT) trial is designed to investigate survival with 300 mg of aspirin daily in the adjuvant setting after completion of surgery and standard chemotherapy with or without radiation therapy for high-risk colon cancer.68 The authors plan to enroll 1587 patients and estimate completion in 2026. The ABC (A Blinded Controlled trial of aspirin as adjuvant breast cancer therapy) trial is designed to evaluate survival with 300 mg of adjuvant aspirin daily for primary invasive human epidermal growth factor receptor 2–negative breast cancer and may provide the earliest data.69 Approximately 3000 patients are enrolled, and trial completion is expected in 2021. Lastly, the ADD-ASPIRIN (Aspirin and disease recurrence and survival after primary therapy in non-metastatic solid tumors) trial is the largest and also expected to be completed in 2026.70 Investigators plan to enroll 11 000 patients to study the effects of aspirin at 100 and 300 mg daily on survival or disease recurrence after standard therapy for 4 tumor types: colorectal, breast, gastroesophageal, and prostate.
While we await the findings of ongoing trials, it is important to put the findings of the ASPREE and ARRIVE trials into context. The recommendation by the 2016 USPSTF to include cancer reduction as part of the benefit for patients 50 to 69 years of age was based on multiple, randomized prospective trials dating back to the adenoma trials published in 2003.48,51 It should also be kept in mind that before ASPREE, the potential benefits of aspirin for cancer prevention were not simply thought to be an extension of its antithrombotic effects. One of the largest trials of CVD prevention analyzed for cancer incidence had a 2 × 2 design that included warfarin in addition to aspirin.71 Whereas aspirin reduced cancer incidence after 3 years and death resulting from cancer after 5 years, there was no benefit from warfarin.
Although the publication of ASPREE has raised new questions about the effects of aspirin on cancer, improved mechanistic understanding regarding different types of cancer and the effects of aspirin during different stages of tumor development may yet explain the discordant human trial data. It is likely that the net effect of aspirin in different cancers depends on both direct effects of the drug on the cancer cells and secondary effects through aspirin-induced platelet inhibition.
Targeting platelet receptors in cancer therapy
The key receptors that mediate platelet-platelet, platelet-leukocyte, and platelet–endothelial cell binding and activation have been interrogated for their mechanistic contribution to cancer metastasis and explored for their potential as anticancer targets.
Platelet P-selectin
Selectins are carbohydrate-binding adhesion proteins that are distinguishable from other calcium-requiring C-type lectins by the presence of an epidermal growth factor–like domain and a series of consensus repeat domains.72 Stored in platelet α granules, platelet P-selectin, or CD62P, recognizes glycan structures primarily in several mucin-like glycoproteins expressed on endothelial cells, leukocytes, and tumor cells. Several high-affinity P-selectin ligands (including sialyl-Lewisx–modified CD24; mucins such as CEA, CA125, and MUC1; CD44 variant; and podocalyxin-like protein 1) have been shown to be variably expressed on cancer cells, including lung, breast, and colon cancer cell lines.73-76 The level of P-selectin ligand expression by cancer cells in culture has been shown to correlate with their metastatic potential. Platelet P-selectin has been shown to play a key role in mediating platelet–tumor cell aggregate formation under physiological conditions of shear flow,77,78 analogous to the role of P-selectin in mediating platelet-leukocyte aggregate formation.79,80 In mouse models, inhibition of P-selectin has been shown to reduce tumor metastasis.81 For instance, injection of P-selectin–knockout mice, as compared with wild-type mice, with colon adenocarcinoma cells resulted in fewer platelet–cancer cell aggregates and diminished homing of tumor cells to the lungs, liver, and kidneys.82 Similar findings have been reported for lung metastasis of melanoma cells in P-selectin–deficient mice.83 Inhibitors targeting platelet P-selectin have recently been translated toward clinical utility.84 Heparin has been shown to block the interaction between P-selectin on platelets and sialyl-Lewisx–bearing mucins and as a result reduce experimental metastasis85,86 ; however, the potential benefit of unfractionated heparin and low molecular weight heparin in survival of cancer patients has not been observed.87-89 This in line with the notion that although anticoagulants play an important role in the management of cancer-associated VTE, the second most common cause of death in patients with cancer,90 the beneficial effects of anticoagulants have not been observed consistently in clinical trials.91 Rather, clinical trials have shown favorable safety and clinical efficacy data for use of the P-selectin antibody crizanlizumab in reducing myocardial damage after percutaneous coronary intervention in patients with non–ST-segment elevation myocardial infarction and in reducing vasoocclusive pain crisis in patients with sickle cell disease ≥16 years of age.92,93 It remains to be seen whether inhibition of P-selectin via antagonists including antibodies or aptamers would be tolerated, safe and efficacious in a chronic setting such as in cancer.
Platelet GPIIbIIIa
The heterodimeric integrin GPIIbIIIa (αIIbβ3, CD41/CD61) is the most abundant and selectively expressed receptor on platelets and megakaryocytes.94,95 Comprising ∼15% by weight of the total protein of the platelet plasma membrane, GPIIbIIIa is com-plexed from the calcium-dependent association of the α and β integrin subunits that require inside-out signaling events to modulate their function in a temporal and spatial manner. This conformational change following platelet activation converts GPIIbIIIa from a low-affinity to a high-affinity receptor for adhesive ligands including fibrinogen, von Willebrand factor, and fibronectin among other components of the blood and extracellular matrix.96,97 GPIIbIIIa plays an essential role in mediating platelet aggregation and adhesion to maintain endothelial barrier function and form hemostatic plugs at sites of vascular injury.98 This shift in the activation state of GPIIbIIIa has been shown to potentiate platelet binding to circulating cells in the bloodstream, including leukocytes, bacteria, and tumor cells.99-101 We and others have shown that platelet GPIIbIIIa mediates platelet–tumor cell binding as well as tumor cell arrest and binding to inflamed endothelial cells under shear flow.4,100,102 Antagonists to platelet GPIIbIIIa or its ligands on tumor cells have been shown to inhibit hematogenous metastasis in mouse models to the same extent as that observed after platelet depletion.99,103,104 However, the long-term use of inhibitors targeting GPIIbIIIa, as would be the case in preventive therapy in cancer, would inherently be coupled with an increased risk of bleeding.105 The well-known physiological role of GPIIbIIIa in hemostasis is evidenced by the bleeding diathesis associated with qualitative or quantitative deficiencies in GPIIbIIIa present in patients with the autosomal recessive disorder Glanzmann thrombasthenia.94 Thus, GPIIbIIIa only represents a suitable target for reducing acute platelet-mediated thrombosis, for instance, during short-term invasive procedures. However, molecular imaging of the active conformation of GPIIbIIIa on platelets has been used for the noninvasive detection of activated platelets in the tumor stroma in vivo106 and thus holds promise for translation to tumor imaging and potentially for targeted radiotherapy.
Platelet CLEC-2
CLEC-2, first identified as a platelet receptor for the snake venom protein rhodocytin, was later shown to be a platelet receptor for podoplanin on tumor cells.16,107 Cross-linking of platelet CLEC-2 by podoplanin signals, through an immunoreceptor tyrosine-based activation motif (ITAM) and via downstream kinases, to induce platelet activation.108 The spatial and temporal expression patterns of podoplanin in select cell types, including fibroblasts, macrophages, T helper cells, and epithelial cells, was then shown to incite platelet activation as a requisite for regulation of vascular/lymphatic development and maintenance of vascular integrity.109 In a pathological setting, the expression of podoplanin by tumor cells has been shown to facilitate hematogenous metastasis by inducing platelet aggregation through CLEC-2.110,111 Studies in select mouse models have indicated a role for platelet CLEC-2 in cancer dissemination and cancer-associated thrombosis. For example, tail vein injection of podoplanin-positive melanoma cells resulted in significantly fewer circulating tumor cells, pulmonary tumor niches, and intratumoral thrombi in mice pretreated with anti–CLEC-2 antibodies than in control mice.110 Furthermore, a synthetic platelet antagonist compound that competes with podoplanin for the same binding site in CLEC-2 also showed antimetastatic properties in vivo.112 Blocking podoplanin seems to exert both antigrowth and antimetastatic effects in vivo; however, podoplanin expression is not specific to cancer cells but instead extends to a wide range of cells in the brain, heart, kidney, lungs, bone, and lymphoid organs, which may be adversely affected by antipodoplanin targeted therapies.108 In contrast, targeting platelet CLEC-2 as a potential anticancer therapy would likely be a safer approach, because although CLEC-2 is predominantly expressed on the surface of platelets and megakaryocytes, it does not seem to play an essential role in hemostasis113,114 ; therefore, blocking CLEC-2 on platelets is unlikely to increase the risk of bleeding in patients with cancer. Furthermore, targeted inhibition of platelet CLEC-2 has been shown to provide benefit in a model of hepatic sterile inflammatory response,115 providing a rationale for therapeutic benefit of blocking platelet CLEC-2 in thromboinflammatory diseases, including acute liver injury, infection, and perhaps cancer-associated thrombosis. Importantly, recent studies have highlighted the fact that low doses of Btk inhibitors including ibrutinib selectively block ITAM-mediated signaling downstream of CLEC-2 relative to the classical collagen receptor GPVI,116,117 providing a rationale for the use of Btk inhibitors in the treatment of diseases that etiologically rely on ITAM-mediated platelet activation.118,119 Along similar lines, tumor cell–expressed galectin-3 has recently been shown to promote ITAM-mediated platelet activation downstream of GPVI, and selectively inhibiting GPVI reduced platelet–tumor cell interaction and tumor metastasis in mouse models.120 The potential targeting of the ITAM-mediated signaling receptors CLEC-2 and GPVI as anticancer therapy will await the outcomes of trials to test the efficacy and safety of targeting these receptors in thromboinflammatory disorders.
Platelet PAR-1
Protease-activated receptors are G-protein–coupled transmem-brane proteins that are triggered by proteolytic cleavage of their N-terminal domains. The protease-activated receptor 1 (PAR-1) is highly expressed on platelets, where its cleavage by thrombin induces strong platelet activation. A variety of tumors, including lung, breast, and ovarian cancers, also express PAR-1, with expression level correlating with adverse prognosis.121 By implication, inhibition of PAR-1 exerts a dual anticancer effect by inhibiting platelets and directly targeting tumor cells.122 Inhibition of PAR-1–G-protein signaling by pepducins or silencing of PAR1 expression with short interference RNA was shown to reduce lung cancer cell migration and reduce growth of lung cancer xenografts.123 In breast cancer models, the pepducin PZ-128 caused >60% inhibition of PAR-1–associated tumor growth and pulmonary metastasis.124,125 On the contrary, an opposite effect of limited tumor growth resulting from PAR-1 signaling has been reported for pancreatic cancer, suggesting that PAR-1 inhibitors may not provide the same benefit in all cancer types.126 Select anti–PAR-1 agents are in clinical trials or have been approved for clinical use as an antiplatelet approach for reducing thrombotic events in patients undergoing cardiac instrumentation127 ; to date, their clinical utility in cancer has yet to be confirmed.
Platelet secondary mediators
Tumors release excessive amounts of nucleosides that contribute to an immunosuppressive microenvironment. Moreover, tumor-derived ADP interacts with purinergic P2Y12 receptors on platelets to induce platelet activation. On the basis of this concept, Cho et al128 demonstrated that pharmacological inhibition of P2Y12 with ticargrelor or transgenic knockout of P2Y12 resulted in >60% decrease in growth of orthotopic ovarian tumors in mice, an effect that was lost when thrombopoiesis was reconstituted in the knockout mice by adoptive transfer.129 Downregulation of tumor cell ectoapyrase, an ADP scavenger, amplified platelet-induced tumor growth, confirming the role of ADP. Although knockdown of P2Y12 messenger RNA in ovarian cancer cells did not affect tumor size, another recent study suggested that inhibition of P2Y12 in pancreatic cancer cell lines abrogated tumor cell proliferation and enhanced tumor sensitivity to chemotherapy.130 Thus, P2Y12 blockade may inhibit the growth of some tumor types via both an antiplatelet effect and direct action on tumor cells. Moreover, inhibition of secondary mediators including ADP would limit their ability to incite GPIIbIIIa activation and translocation of P-selectin on platelets, both of which are known to facilitate hematogenous metastasis. As a proof of concept, depletion of apoptosis signal–regulating kinase 1, a protein involved in ADP-related intraplatelet signaling, attenuated pulmonary metastasis in mice.131 Although P2Y12 antagonists are therapeutic options for disrupting tumor growth and metastasis from a mechanistic standpoint, whether their benefit would outweigh the risk of bleeding associated with P2Y12 would have to be considered in chronic use as a cancer therapy. Large trials for CVD reduction with P2Y12 inhibitors have not shown an association of either clopidogrel or ticagrelor with cancer outcomes.132-134 A large trial comparing prasugrel with clopidogrel reported a statistically significant increase of colon cancer with prasugrel.135 However, the incidence was small (13 vs 4 cases) in a trial of >13 000 patients. A more in-depth review by the US Food and Drug Administration did find an excess of 28 total cancers among patients assigned prasugrel; however, the conclusion was that this represented a very low incidence, which was not concerning and required more study.136,137 More recent clinical data, including post hoc analysis of the TRILOGY-ACS study,138 have failed to show an association between prasugrel and cancer, and a comprehensive review did not find a positive or negative association of P2Y12 inhibitors with cancer.139,140
Lastly, receptor-mediated platelet activation stimulates the release of platelet-derived growth factors, which are known to drive tumor cell growth and differentiation.141 Several bioactive proteins in the platelet secretome have been shown to drive proliferative and survival signals in tumors including platelet-derived growth factor, vascular endothelial growth factor, epithelial growth factor, transforming growth factor β, and cytokines. Although the components of the platelet secretome may represent biomarkers of platelet–tumor cell crosstalk, their essential role in maintaining homeostasis, including angiogenesis, tissue regeneration during wound repair, and barrier function, preclude several components of the platelet secretome as druggable targets for management or prevention of cancer.
Platelet count reduction for cancer therapy
The prevalence and severity of cancer-associated thrombocytosis vary with type of cancer and stage of malignancy.22 For example, <5% of patients with breast cancer were found to have high platelet counts, compared with 25% to 55% of patients with CRC and lung and ovarian cancers, with higher count associated with worse cancer prognosis.19-21,23 Although risk of paraneoplastic thrombocytosis is higher in advanced cancer than in early cancer, a recent historical cohort study showed that in cancer-naïve individuals age ≥40 years, high platelet count is an independent predictor of 1-year risk of new cancer diagnosis.142 We recently demonstrated that temporal platelet count information from a cohort of >10 000 patients with lung, prostate, and colon cancers improved prognostication compared with a single time point at the time of treatment.143 However, the mechanistic basis of paraneoplastic thrombocytosis is not fully established. Nevertheless, Stone et al21 demonstrated that ovarian tumor–derived interleukin-6 (IL-6) stimulated hepatic thrombopoietin (TPO) release, which promoted megakaryopoiesis and subsequent thrombopoiesis. The authors demonstrated that blocking IL-6 and TPO production with a short interference RNA led to normalization of platelet counts in an animal model of ovarian cancer. A significant reduction in tumor growth was observed in mouse models when IL-6 was targeted with siltuximab (an anti–IL-6 antibody) and combined with paclitaxel (a common chemotherapeutic agent for epithelial ovarian cancer). From such work, it is conceivable that drugs reducing platelet counts or platelet activation might attenuate cancer progression and improve overall outcomes in cancer.
Considering that the hemostatic function of platelets is largely preserved, even when platelet counts drop to as low as 10 × 103/μL from the normal adult count of 150 to 450 × 103/μL of blood,144 we questioned whether reducing platelet counts within the hemostatic window represents a safe anticancer approach. Using antisense oligonucleotides that selectively silence hepatic TPO, Shirai et al145 successfully reduced bone marrow megakaryocyte density and decreased circulating platelet counts by 50% in both murine and primate models. The lowered but hemostatic platelet counts resulted in lower levels of circulating vascular endothelial growth factor, reduced primary tumor growth and vascularization, reduced pulmonary metastasis, and increased survival in a mouse model of spontaneous metastatic breast cancer. These data lend credence to the concept that reducing platelet count within the hemostatic range may constitute a safer anticancer alternative than systemically targeting platelet receptors or activation pathways. However, cautious optimism should be exercised in the development of anti-TPO agents as adjuvant therapy in cancer, because they would not be well suited for those patients who receive myelotoxic treatments and may not be used in immune- or infection-associated thrombocytopenia, which could be exacerbated by anti-TPO platelet reduction.146
Discussion
Because platelets within the normal physiological range can support tumor progression, use of antiplatelet therapies has been posited as a strategy to interdict or prevent the signaling events that drive cancer growth and metastasis. The use of antiplatelet agents for primary cancer prevention implies long-term administration, which cumulatively increases the risk of bleeding; therefore, this would favor approaches that do not compromise the hemostatic level or function of platelets, such as targeting platelet CLEC-2. Additional clinical scenarios where benefit for risk may be favorable include the administration of antiplatelet agents as shorter-term adjuvants to radiotherapy or surgery. Newer clinical trials are reminders of the challenges that remain: to establish safe antiplatelet approaches to improve cancer outcomes and to understand the complex progression of pathological processes that are promoted by platelets.
Acknowledgments
The authors thank Joseph Shatzel, András Gruber, Garth Tormoen, Monica Burdick, and Annachiara Mitrugno for insightful discussions.
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01HL101972 and R01HL144113), the Oregon Health & Science University/Oregon State University Cancer Prevention and Control Initiative, and the Knight Cancer Institute Cancer Early Detection Advanced Research Center.
Authorship
Contribution: D.L.T., S.T.Y., C.D.W., and O.J.T.M. wrote and approved the final version of manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Owen J. T. McCarty, Department of Biomedical Engineering, Oregon Health & Science University, 3303 S Bond Ave, Mail Code CH13B, Portland, OR 97239; e-mail: mccartyo@ohsu.edu.