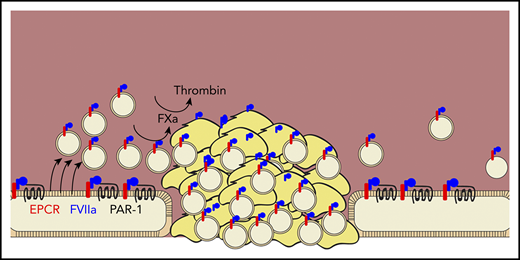
In this issue of Blood, 1 describe a new hemostatic mechanism of recombinant factor VIIa (rFVIIa), a bypassing agent used to prevent bleeding in patients with hemophilia (see figure).2 Although it has been in use for more than 30 years, researchers are still discovering the mechanisms by which rFVIIa promotes hemostatic clot formation. The canonical function of FVIIa is to bind its cofactor tissue factor (TF) and activate coagulation factor X (FX), the enzyme responsible for production of thrombin.3 However, the high concentrations of rFVIIa that are used clinically allow it to have TF-independent functions. In 1990, it was shown that therapeutic concentrations of rFVIIa can promote FX activation independent of TF,4 and in 1997, it was shown that rFVIIa can bind to the surface of activated platelets and promote thrombin generation in the absence of TF.5 This TF-independent function requires the presence of a procoagulant, phosphatidylserine (PS)-containing membrane surface to which FVIIa and FX can localize. In the current study, Das et al1 show that FVIIa can directly generate this surface in the form of PS-containing extracellular vesicles (EVs) released by endothelial cells.
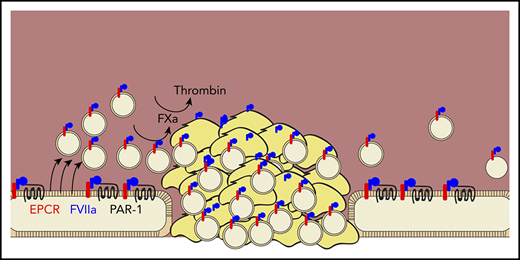
Das et al demonstrate that rFVIIa (blue) promotes hemostasis by binding to the endothelial protein C receptor (red), promoting the cleavage of protease-activated receptor 1 (black line), and inducing the release of procoagulant extracellular vesicles (tan circles) from endothelial cells. These vesicles localize into growing clots, bind factor VIIa, and promote the activation of factor Xa and thrombin.
Das et al demonstrate that rFVIIa (blue) promotes hemostasis by binding to the endothelial protein C receptor (red), promoting the cleavage of protease-activated receptor 1 (black line), and inducing the release of procoagulant extracellular vesicles (tan circles) from endothelial cells. These vesicles localize into growing clots, bind factor VIIa, and promote the activation of factor Xa and thrombin.
This group has previously shown that rFVIIa binds the endothelial protein C receptor (EPCR), which promotes the cleavage of protease activated receptor-1 (PAR-1) and results in a cytoprotective phenotype.6 Here, they extended this previous work by showing that PAR-1 cleavage causes the release of EVs, specifically from endothelial cells, both in vitro and in vivo. These EVs express PS and promote TF-independent FX activation by rFVIIa. They also promote thrombin activation by FXa and its cofactor factor Va. EV production required EPCR and was mediated through cleavage of PAR-1 at R41Q, the same mechanism by which FVIIa induces cytoprotection.7 In a saphenous vein injury model, EVs localize to the injury site and support hemostatic clot formation, as evidenced by decreased blood loss and clot time and increased thrombin–antithrombin complex. This was true in hemophilia A mice and in wild-type mice treated with an antibody to deplete platelets, suggesting that the hemostatic effect of EVs can compensate for the loss of platelets.
This study raises several questions that can drive future research endeavors, both related to the pharmacologic properties of rFVIIa and to normal hemostasis. For example, what properties of endothelial-derived EVs allow them to promote hemostasis? The authors identified EVs from endothelial cells, platelets, monocytes, and red blood cells, but rFVIIa only induced an increase in endothelial EVs and only promoted hemostasis when endothelial EVs were present. This suggests that endothelial EVs possess unique lipid and/or protein components that confer their functional properties, such as their ability to localize to an injury site and promote hemostasis. The authors identified EPCR, vascular endothelial (VE)-cadherin, and PS as components of the vesicles and showed that PS is essential for promoting FXa and thrombin activation. Does EPCR also promote FXa activation by binding FVIIa? Does VE-cadherin serve as an adhesive receptor that binds exposed subendothelium and localizes EVs to the injury? Are there other key lipids or proteins present? A complete characterization of these EVs would help us to better understand rFVIIa function.
The model presented here provides a starting point to study the potential for endothelial EVs to participate in normal hemostasis as well. That is because other enzymes may also induce release of EVs from endothelial cells. Thrombin, for example, activates PAR-1 by cleavage at Arg41.8 Does thrombin generated at an injury site function through a similar, although likely EPCR-independent, mechanism to release EVs and promote its own activation? Similarly, at physiologic FVIIa concentrations, does the TF/FVIIa complex promote EV formation, or is this activity of FVIIa only relevant under therapeutic conditions?
Finally, the authors also demonstrated that rFVIIa causes a procoagulant surface to form on the endothelial cell itself. rFVIIa-treated endothelial cells expressed PS and supported FXa and thrombin activation. Is this endothelial cell activation localized to the injury site or is it systemic? If rFVIIa can induce a procoagulant surface in any endothelial cell that expresses both EPCR and PAR-1, then what mechanisms prevent these cells from promoting systemic clot formation, and could those mechanisms be exploited for future anticoagulant therapies? There is still much to learn about the control of hemostasis and thrombosis, and the role of endothelial cells and endoethelial EVs in this process. This study has expanded our knowledge base and opened up many new avenues of research by showing that rFVIIa is more than just a FX activator. It is also a potent activator of endothelial cells and can provide the membrane surface required for that FXa to function.
Conflict-of-interest disclosure: The author declares no competing financial interests