

Key Points
FVIIa treatment induces the release of EVs from the endothelium into circulation via FVIIa-EPCR-PAR1–mediated biased signaling.
FVIIa-EEVs exhibit higher procoagulant activity and are capable of correcting bleeding associated with platelet deficiency or hemophilia.

Abstract
Recombinant factor FVIIa (rFVIIa) is used as a hemostatic agent to treat bleeding disorders in hemophilia patients with inhibitors and other groups of patients. Our recent studies showed that FVIIa binds endothelial cell protein C receptor (EPCR) and induces protease-activated receptor 1 (PAR1)-mediated biased signaling. The importance of FVIIa-EPCR-PAR1–mediated signaling in hemostasis is unknown. In the present study, we show that FVIIa induces the release of extracellular vesicles (EVs) from endothelial cells both in vitro and in vivo. Silencing of EPCR or PAR1 in endothelial cells blocked the FVIIa-induced generation of EVs. Consistent with these data, FVIIa treatment enhanced the release of EVs from murine brain endothelial cells isolated from wild-type (WT), EPCR-overexpressing, and PAR1-R46Q–mutant mice, but not EPCR-deficient or PAR1-R41Q–mutant mice. In vivo studies revealed that administration of FVIIa to WT, EPCR-overexpressing, and PAR1-R46Q–mutant mice, but not EPCR-deficient or PAR1-R41Q–mutant mice, increased the number of circulating EVs. EVs released in response to FVIIa treatment exhibit enhanced procoagulant activity. Infusion of FVIIa-generated EVs and not control EVs to platelet-depleted mice increased thrombin generation at the site of injury and reduced blood loss. Administration of FVIIa-generated EVs or generation of EVs endogenously by administering FVIIa augmented the hemostatic effect of FVIIa. Overall, our data reveal that FVIIa treatment, through FVIIa-EPCR-PAR1 signaling, releases EVs from the endothelium into the circulation, and these EVs contribute to the hemostatic effect of FVIIa.
Introduction
Recombinant factor VIIa (rFVIIa) is widely used to treat bleeding disorders in hemophilia patients with inhibitors and bleeding caused by a wide array of clinical scenarios.1-3 It is generally believed that rFVIIa provides the therapeutic hemostatic effect in hemophilia patients via a platelet-dependent mechanism.4 Studies from us and others showed that factor VIIa (FVIIa), whose primary function is to initiate blood coagulation by binding to tissue factor (TF) following the vascular injury,5 also binds anticoagulant cofactor endothelial cell protein C receptor (EPCR).6-8 EPCR is a receptor for anticoagulant protein C or activated protein C (APC).9 It plays a crucial role in the protein C anticoagulant pathway by promoting the activation of protein C by the thrombin-thrombomodulin complex.10 Recent studies establish that EPCR plays a key role in supporting APC-mediated cytoprotective signaling.11-13 A series of studies from our laboratory showed that EPCR also supports FVIIa-induced cytoprotective signaling.14-17 Although both APC and FVIIa induce EPCR-dependent cytoprotective signaling through activation of PAR1-mediated cell signaling, important differences exist between APC and FVIIa in their mode of action. For example, APC was shown to cleave PAR1 preferentially at a noncanonical Arg46 site to induce cytoprotective signaling.18,19 In contrast, FVIIa-mediated anti-inflammatory signaling requires the cleavage of PAR1 at the canonical Arg41 site.17 It is unknown at present whether EPCR-FVIIa-PAR1 signaling, either directly or indirectly, affects the hemostatic process.
Cells release extracellular vesicles (EVs) constitutively or in response to injury, stress, inflammation, or other pathophysiologic conditions.20 At present, 3 distinct populations of EVs have been described based on their biogenesis and size: exosomes, microvesicles (microparticles), and apoptotic bodies.21,22 However, in the absence of consensus on specific markers of EV subtypes and difficulties in identifying them accurately, it was recommended to use the generic term EV to describe all subtypes of EVs.23 EVs are readily found in the blood of healthy humans, and their levels are elevated in a variety of diseases.22,24,25 The majority of EVs detected in the blood of healthy subjects are derived from platelets and red blood cells (RBCs), and only a small fraction of EVs are released from endothelial cells.24,26-28 Various pathological conditions, including coronary syndrome,25,29 antiphospholipid syndrome,30 and sickle cell disease,31 were found to increase the levels of endothelial EVs (EEVs). Several studies showed that endothelial cells release EVs in response to inflammatory stimuli.30,32-35 The procoagulant/prothrombotic effect of EEVs appears to come from TF, as many inflammatory stimuli induce TF expression in endothelial cells and EEVs released from the activated endothelial cells carry TF on their surface.30,32,34 At present, there is no evidence for EEVs released from unperturbed endothelium to support the coagulation.
In the present study, we demonstrate that FVIIa induces the release of EVs from endothelial cells, both in vitro and in vivo settings. FVIIa-induced EEVs release is dependent on EPCR and PAR1. We also show that FVIIa-derived EEVs are devoid of TF but exhibit higher phosphatidylserine (PS)-dependent procoagulant activity than EVs generated constitutively. In vivo studies show that administration of FVIIa-generated EEVs corrects the bleeding in platelet-depleted and hemophilia A mice without causing systemic activation of coagulation. These findings are novel and are of great clinical significance as FVIIa is used as a therapeutic drug for treating bleeding-related disorders.
Materials and methods
See supplemental Materials and methods (available on the Blood Web site) for additional details.
Mice
Endothelial cells
Primary human umbilical vein endothelial cells (HUVECs) were obtained from Lonza (Walkersville, MD) and cultured in endothelial basal medium supplemented with endothelial-specific growth factors. Mouse brain endothelial cells from wild-type (WT) and transgenic mice were isolated and cultured as described in our recent publication.17
Silencing of EPCR, PAR1, PAR2, or PAR4
HUVECs were transfected with small interfering RNA (siRNA) specific for EPCR, PAR1, PAR2, or PAR4. As controls, scrambled nucleotide of the sequences of the above siRNAs were used. The transfection was carried out using Lipofectamine RNAiMAX or X-tremeGENE siRNA transfection reagent (Sigma-Aldrich) in a serum-free medium.
Isolation and characterization of EVs
EVs from cell culture supernatants and mouse blood were isolated by a procedure described earlier, with a few minor modifications.38-40 This isolation procedure yields EVs that are generally referred to as microvesicles or microparticles in most of the earlier publications in the field. The number of EVs and their diameter were measured utilizing nanoparticle tracking analysis (NTA) with NanoSight 300 (Malvern Panalytical).
FXa generation and prothrombinase assays
The rate of factor X (FX) and prothrombinase activation on intact cells and EVs was measured in a chromogenic assay, as described earlier.41,42
Animal studies
To investigate the role of EPCR-PAR1 signaling in FVIIa-induced EV generation, WT, EPCR-KO, EPCR-OX, PAR1-R41Q, and PAR1-R46Q mice were injected with saline or FVIIa (0.25 mg/kg in 100 µL). Two hours following rFVIIa administration, blood was drawn into citrate anticoagulant, and EVs were isolated from platelet-free plasma.
To investigate the potential effect of FVIIa-induced EVs on hemostasis in vivo, we first depleted platelets in WT mice by administering platelet depletion antibody (CD42b monoclonal antibody [mAb], 1 mg/kg, Emfret, Germany). Following the antibody administration, mice were injected with control EVs or FVIIa-generated EVs (1 × 109 EVs per mouse). Control EVs and FVIIa-generated EVs were prepared by treating cultured endothelial cells with either a control vehicle or FVIIa (100 nM) for 24 hours, respectively, and isolating EVs from the supernatant medium. After 5 minutes, mice were subjected to the saphenous vein incision. Blood coming from the incision site was absorbed onto Kim wipes to measure blood loss, as described in our earlier publication.43 A small aliquot of blood from the injury site was also collected into citrate anticoagulant to measure thrombin-antithrombin (TAT) complex levels as an index for thrombin generation.
In a second model, FVIII−/− mice were administered with control EVs or FVIIa-generated EVs from HUVECs (1.5 × 109 EVs per mouse) and then treated with FVIIa (0.25 mg/kg). Mice were subjected to the saphenous vein injury, and the average time to achieve hemostasis and total blood loss were determined as described earlier.43 Alternatively, FVIII−/− mice were pretreated with FVIIa (0.25 mg/kg) for the endogenous generation of EVs, and 2 hours later, mice were subjected to the saphenous vein incision with or without administering a second dose of FVIIa (0.25 mg/kg) before the injury. A dose of 0.25 mg/kg FVIIa was chosen here, as this dose was sufficient to induce EPCR-PAR1–mediated signaling but had no hemostatic effect in hemophilia mice.15,17,44 Since 3 to 10 mg/kg rFVIIa is typically required to correct the bleeding disorder in FVIII−/− mice,44,45 the dose of 0.25 mg/kg rFVIIa used in the present study might be considered low for a murine model. Thrombin generated at the wound site was measured by collecting the blood from the wound site and measuring TAT levels.
Statistical analysis
All experiments described in this report were repeated at least 3 times, and 6 to 10 mice were used for each experimental group. Data are presented as mean ± standard error of the mean (SEM). Statistically significant differences between the 2 groups were analyzed by Student t test. One-way analysis of variance, followed by Bonferroni’s post hoc multiple comparison tests, were used to determine the statistical significance among more than 2 groups.
Results
FVIIa induces EV generation from endothelial cells
In initial experiments to investigate the effect of FVIIa on EV generation in endothelial cells, endothelial cell surface membrane was labeled with the cell-impermeable fluorescent dye PKH67 (Figure 1A) and exposed to FVIIa for varying times. Measurement of EV generation by monitoring the fluorescence intensity of EVs isolated from cell supernatants showed that FVIIa treatment markedly increased the generation of EVs from HUVECs in a time-dependent manner (Figure 1B). Next, we analyzed the generation of EVs in unlabeled HUVECs by NTA. Treatment of HUVECs with FVIIa (100 nM) for 24 hours increased the generation of EVs by four- to fivefold over the control vehicle treatment (Figure 1C). FVIIa-induced EVs appear to be homogenous in their size distribution, with a mean diameter of 149.1 ± 4.4 nm (Figure 1D). The size distribution of EVs generated in the control treatment was heterogeneous, with a mean diameter of 184.1 ± 16.3 nm (Figure 1D). Analysis of EVs by immunoblotting showed that FVIIa-induced EVs contained EPCR and vascular-endothelial (VE)-cadherin, endothelial cell proteins embedded in the plasma membrane (Figure 1E-G). Determination of FVIIa levels in EVs by immunoblot analysis (supplemental Figure 1) or FVIIa-specific coagulation assay (detection limit, 0.5 ng/mL) showed no detectable amount of FVIIa in either control- or FVIIa-derived EVs. However, FVIIa was readily detectable by mass spectrometry analysis of FVIIa-derived EVs (data not shown).
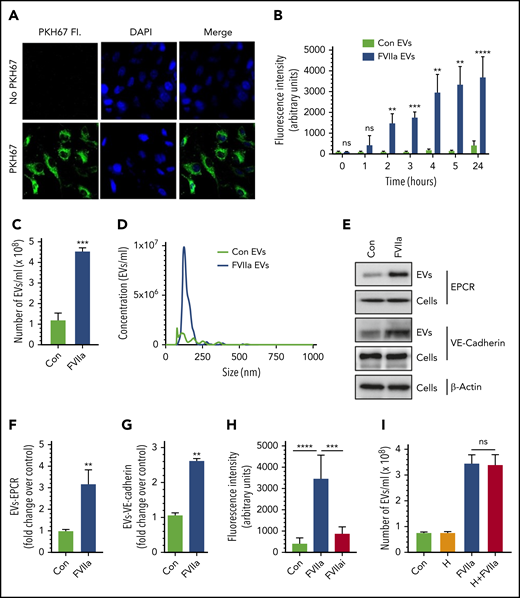
FVIIa induces the release of EVs from endothelial cells. (A) HUVECs were incubated with PKH67 dye (20 nM) for 30 minutes at 37°C in a serum-free medium. The cells were washed 3 times with Hanks balanced salt solution, fixed with 4% paraformaldehyde, and stained with 4′,6-diamidino-2-phenylindole (DAPI) for 30 minutes. The labeled cells were subjected to fluorescence (Fl.) microscopy. (B) PKH67-labeled cells were serum starved for 1 hour, followed by treatment with FVIIa (100 nM) or a control (Con) vehicle for varying times. EVs isolated from the supernatant at the indicated times were quantified by measuring the fluorescence intensity of PKH67 dye. (C) Cells grown onto 6-well culture dishes were treated with a control vehicle or FVIIa (100 nM) for 24 hours. EVs isolated from culture supernatants were quantified by NTA NanoSight. (D) A spectrum showing the diameter range of EVs as analyzed by NTA NanoSight. (E) HUVECs were treated with a control vehicle or FVIIa (100 nM) for 24 hours. EVs isolated from cell supernatants and cell lysates were subjected to immunoblotting to probe endothelial cell surface markers, EPCR, and VE-cadherin (loading, EVs from 1 × 106 cells; cell lysates from 0.5 × 105 cells). (F-G) Band intensities were quantified by densitometric analysis. (H) PKH67-labeled cells were treated with a control vehicle, FVIIa (100 nM) or FVIIai (100 nM) for 24 hours. EVs were quantified by fluorescence measurement. (I) HUVECs were incubated with hirudin (H; 4U/mL) for 1 hours, followed by FVIIa (100 nM) for 24 hours. EV generation was quantified by NTA NanoSight. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, not statistically significantly different.
FVIIa induces the release of EVs from endothelial cells. (A) HUVECs were incubated with PKH67 dye (20 nM) for 30 minutes at 37°C in a serum-free medium. The cells were washed 3 times with Hanks balanced salt solution, fixed with 4% paraformaldehyde, and stained with 4′,6-diamidino-2-phenylindole (DAPI) for 30 minutes. The labeled cells were subjected to fluorescence (Fl.) microscopy. (B) PKH67-labeled cells were serum starved for 1 hour, followed by treatment with FVIIa (100 nM) or a control (Con) vehicle for varying times. EVs isolated from the supernatant at the indicated times were quantified by measuring the fluorescence intensity of PKH67 dye. (C) Cells grown onto 6-well culture dishes were treated with a control vehicle or FVIIa (100 nM) for 24 hours. EVs isolated from culture supernatants were quantified by NTA NanoSight. (D) A spectrum showing the diameter range of EVs as analyzed by NTA NanoSight. (E) HUVECs were treated with a control vehicle or FVIIa (100 nM) for 24 hours. EVs isolated from cell supernatants and cell lysates were subjected to immunoblotting to probe endothelial cell surface markers, EPCR, and VE-cadherin (loading, EVs from 1 × 106 cells; cell lysates from 0.5 × 105 cells). (F-G) Band intensities were quantified by densitometric analysis. (H) PKH67-labeled cells were treated with a control vehicle, FVIIa (100 nM) or FVIIai (100 nM) for 24 hours. EVs were quantified by fluorescence measurement. (I) HUVECs were incubated with hirudin (H; 4U/mL) for 1 hours, followed by FVIIa (100 nM) for 24 hours. EV generation was quantified by NTA NanoSight. *P < .05; **P < .01; ***P < .001; ****P < .0001. ns, not statistically significantly different.
Both control- and FVIIa-generated EVs were devoid of Alix, an exosome marker protein (supplemental Figure 2), indicating that the EVs isolated in our study only represent microvesicles and exclude exosomes. Analysis of exosomes, isolated by centrifugation of the conditioned media at 100 000g for 1 hour, showed that FVIIa treatment did not affect the release of exosomes (supplemental Figure 2). Since endothelial cells vesiculate when activated or undergo apoptosis,46,47 we investigated the possibility of FVIIa-derived EVs originating from endothelial cells undergoing apoptosis. FVIIa-treated cells were fully viable, as determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-dimethyltetrazolium bromide assay (supplemental Figure 3A). There was no evidence for FVIIa treating cells undergoing apoptosis, as PARP1 immunoblot analysis showed no cleaved PARP1 in FVIIa-treated cells (supplemental Figure 3B). Moreover, our earlier studies showed that FVIIa suppresses endothelial cell activation.16,17
Like HUVECs, primary human aortic endothelial cells and murine brain endothelial cells (bEND.3) also showed enhanced EV generation upon exposure to FVIIa (supplemental Figure 4A-B). FVIIa’s proteolytic activity was necessary for FVIIa to induce EV generation, as proteolytically inactive FVIIa (FVIIai) had no significant effect on EV generation (Figure 1H). It may be pertinent to note here that FVIIai, similar to FVIIa, binds EPCR.48,49 Undetectable levels of thrombin that could potentially generate in the cell system upon the addition of FVIIa was not responsible for increased EV generation, as a thrombin-specific inhibitor, hirudin, did not affect FVIIa-induced EV generation in endothelial cells (Figure 1I).
FVIIa-mediated EV generation: EPCR and PAR1 dependency
To investigate the role of FVIIa-EPCR-PAR1 signaling in FVIIa-induced EV generation in endothelial cells, EPCR expression in HUVECs was silenced by EPCR-specific siRNA (Figure 2A). EPCR silencing completely attenuated the FVIIa-induced EV generation (Figure 2B). Additional experiments showed that blocking FVIIa binding to EPCR with EPCR-blocking mAb abolished FVIIa’s ability to release EVs from endothelial cells (Figure 2C). Next, to determine the contribution of PAR1 in the FVIIa-induced EVs production, PAR1 expression in HUVECs was silenced by siRNA (Figure 2D), or PAR1 activation was blocked by ATAP2 antibody (Figure 2F) before the cells were exposed to FVIIa. PAR1 silencing completely prevented the FVIIa-induced EVs generation (Figure 2E). Similarly, the treatment of HUVECs with the PAR1 antibody fully attenuated the FVIIa-induced EV generation (Figure 2F). In contrast to the above data, neither TF antibody nor PAR2 silencing had any effect on FVIIa-induced EV release from endothelial cells (Figure 2G-I). Similarly, PAR4 silencing had no effect on FVIIa release of EVs from endothelial cells (supplemental Figure 5A-B).
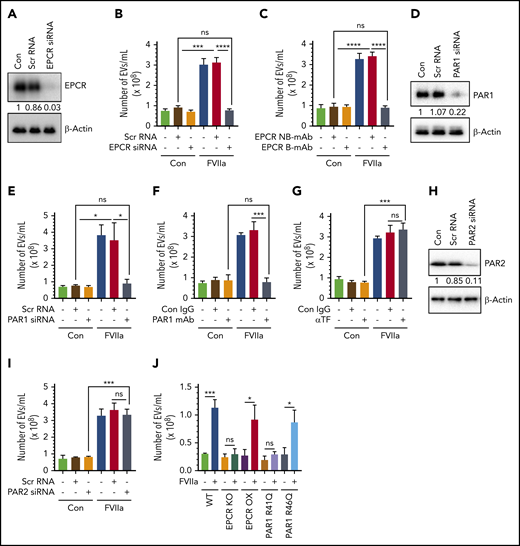
FVIIa-mediated EV generation from HUVECs is dependent on the EPCR-PAR1 signaling axis. (A) HUVECs were transfected with scrambled RNA (Scr RNA; 200 nM) or EPCR-specific siRNA (EPCR siRNA; 200 nM) using Lipofectamine RNAiMAX reagent. After 48 hours, cells were lysed, and EPCR expression was analyzed by western blotting using a human EPCR–specific antibody. (B) Scrambled RNA and EPCR siRNA-transfected cells were treated with control vehicle or FVIIa (100 nM). Twenty-four hours later, cell supernatants were collected, and EVs were isolated and quantified by NTA NanoSight. (C) HUVECs were preincubated with EPCR blocking antibody (EPCR B-mAb; JRK1494; 100 µg/mL) or nonblocking control antibody (EPCR NB-mAb; JRK1500; 100 µg/mL) for 1 hour prior to the addition of FVIIa (100 nM). EVs released from these cells after 24 hours were quantified by NTA NanoSight. (D) HUVECs were transfected with scrambled siRNA (100 nM) or PAR1-specific siRNA (PAR1 siRNA; 100 nM) in a similar approach as mentioned in panel A, and PAR1 knockdown was verified by western blotting with human PAR1–specific antibody. (E) Control and PAR1 siRNA-transfected cells were treated with FVIIa (100 nM) or control vehicle for 24 hours. EVs isolated from cell supernatants were quantified using NTA NanoSight. (F) HUVECs were pretreated with PAR1-specific blocking antibodies (PAR1 mAb; ATAP2; 25 µg/mL) or control immunoglobulin G (IgG; 25 µg/mL) for 1 hour before the addition of FVIIa (100 nM) or control vehicle. After 24 hours, EVs in cell supernatants were quantified by NTA NanoSight. (G) HUVECs were treated with control IgG (Con IgG; 10 µg/mL) or TF neutralizing antibody (αTF; 10 µg/mL) for 1 hour followed by FVIIa (100 nM) or a control vehicle. After 24 hours, EVs released into cell supernatant were quantified by NTA NanoSight. (H) HUVECs were transfected with scrambled siRNA (100 nM) or PAR2-specific siRNA (PAR2 siRNA, 100 nM). After 48 hours of transfection, PAR2 expression in cells was analyzed by western blotting. (I) PAR2 siRNA or scrambled RNA–transfected or control cells were treated with control vehicle or FVIIa (100 nM) for 24 hours, and EVs released in the supernatant culture medium were quantified by NTA NanoSight. (J) Murine brain endothelial cells were isolated from WT C57BL/6J (WT), EPCR knockout (EPCR-KO), EPCR-overexpressing (EPCR-OX), PAR1-R41Q mutant (PAR1-R41Q), and PAR1-R46Q–mutant (PAR1-R46Q) mice as mentioned in “Materials and methods.” Confluent monolayers of cells were treated with FVIIa (100 nM) or a control vehicle for 24 hours. EVs, isolated from the cell supernatants, were quantified by NTA NanoSight. Data are presented as mean ± SEM from 3 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
FVIIa-mediated EV generation from HUVECs is dependent on the EPCR-PAR1 signaling axis. (A) HUVECs were transfected with scrambled RNA (Scr RNA; 200 nM) or EPCR-specific siRNA (EPCR siRNA; 200 nM) using Lipofectamine RNAiMAX reagent. After 48 hours, cells were lysed, and EPCR expression was analyzed by western blotting using a human EPCR–specific antibody. (B) Scrambled RNA and EPCR siRNA-transfected cells were treated with control vehicle or FVIIa (100 nM). Twenty-four hours later, cell supernatants were collected, and EVs were isolated and quantified by NTA NanoSight. (C) HUVECs were preincubated with EPCR blocking antibody (EPCR B-mAb; JRK1494; 100 µg/mL) or nonblocking control antibody (EPCR NB-mAb; JRK1500; 100 µg/mL) for 1 hour prior to the addition of FVIIa (100 nM). EVs released from these cells after 24 hours were quantified by NTA NanoSight. (D) HUVECs were transfected with scrambled siRNA (100 nM) or PAR1-specific siRNA (PAR1 siRNA; 100 nM) in a similar approach as mentioned in panel A, and PAR1 knockdown was verified by western blotting with human PAR1–specific antibody. (E) Control and PAR1 siRNA-transfected cells were treated with FVIIa (100 nM) or control vehicle for 24 hours. EVs isolated from cell supernatants were quantified using NTA NanoSight. (F) HUVECs were pretreated with PAR1-specific blocking antibodies (PAR1 mAb; ATAP2; 25 µg/mL) or control immunoglobulin G (IgG; 25 µg/mL) for 1 hour before the addition of FVIIa (100 nM) or control vehicle. After 24 hours, EVs in cell supernatants were quantified by NTA NanoSight. (G) HUVECs were treated with control IgG (Con IgG; 10 µg/mL) or TF neutralizing antibody (αTF; 10 µg/mL) for 1 hour followed by FVIIa (100 nM) or a control vehicle. After 24 hours, EVs released into cell supernatant were quantified by NTA NanoSight. (H) HUVECs were transfected with scrambled siRNA (100 nM) or PAR2-specific siRNA (PAR2 siRNA, 100 nM). After 48 hours of transfection, PAR2 expression in cells was analyzed by western blotting. (I) PAR2 siRNA or scrambled RNA–transfected or control cells were treated with control vehicle or FVIIa (100 nM) for 24 hours, and EVs released in the supernatant culture medium were quantified by NTA NanoSight. (J) Murine brain endothelial cells were isolated from WT C57BL/6J (WT), EPCR knockout (EPCR-KO), EPCR-overexpressing (EPCR-OX), PAR1-R41Q mutant (PAR1-R41Q), and PAR1-R46Q–mutant (PAR1-R46Q) mice as mentioned in “Materials and methods.” Confluent monolayers of cells were treated with FVIIa (100 nM) or a control vehicle for 24 hours. EVs, isolated from the cell supernatants, were quantified by NTA NanoSight. Data are presented as mean ± SEM from 3 independent experiments. *P < .05; **P < .01; ***P < .001; ****P < .0001.
In additional studies, murine brain endothelial cells isolated from WT, EPCR-KO, EPCR-OX, PAR1-R41Q, and PAR1-R46Q–mutant mice were exposed to FVIIa for 24 hours, and EVs in the conditioned media were isolated and analyzed by NTA. FVIIa treatment increased the generation of EVs approximately threefold in endothelial cells isolated from WT, EPCR-OX, and PAR1-R46Q–mutant mice, but not EPCR-KO or PAR1-R41Q–mutant mice (Figure 2J).
The hemostatic potential of FVIIa-induced EVs
To investigate the hemostatic potential of EVs, EVs isolated from control vehicle- and FVIIa-treated HUVECs were evaluated for their ability to support FVIIa-catalyzed activation of FX and FXa/FVa-mediated activation of prothrombin. When compared with control EVs, FVIIa-generated EVs enhanced FX and prothrombin activation by approximately six- to ninefold (Figure 3A-B). Preincubation of EVs with annexin V completely abolished the increased activation of FX and prothrombin supported by FVIIa-generated EVs (Figure 3A-B). In contrast, pretreatment of EVs with TF neutralizing antibodies had no effect on the enhanced rate of FX activation. The above data do not reveal whether the increased activation of FX and prothrombin by FVIIa-induced EVs solely reflects the increased number of EVs generated upon FVIIa treatment of endothelial cells or FVIIa-generated EVs per se possess higher procoagulant activity. To investigate the above possibility, we analyzed the rate of FX and prothrombin activation using an equal number of EVs isolated from control- and FVIIa-treated endothelial cells. Even after normalization of EV number, FVIIa-generated EVs exhibited an approximately threefold higher capacity to support FX and prothrombin activation as compared with control EVs (Figure 3C-D). Similar results were obtained with EVs generated from endothelial cells from other vascular beds (supplemental Figure 6A-D). In additional studies, we investigated whether FVIIa-derived EVs also support protein C activation by thrombin at an enhanced rate compared with control EVs. After normalization, control- and FVIIa-generated EVs contained similar thrombomodulin levels and supported protein C activation at similar rates (supplemental Figure 7A-B).
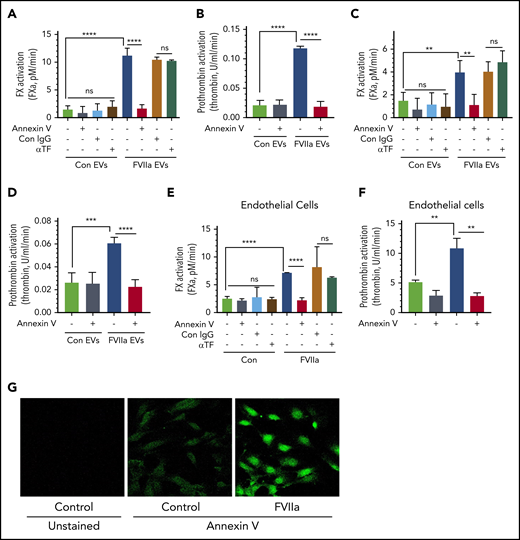
FVIIa-induced EVs exhibit higher procoagulant activity in vitro, and this is dependent on anionic phospholipids and independent of TF. (A) EVs released from HUVECs upon control vehicle or FVIIa treatment (100 nM for 24 hours) were isolated and suspended into equal amounts of a calcium-containing buffer. EVs were incubated with either annexin V (400 nM), TF neutralizing antibodies (10 µg/mL), or control IgG (10 µg/mL) for 1 h. After that, FVIIa (10 nM) was added to the EV suspension, and FX (175 nM) was added 5 minutes later. The rate of FX activation was measured in a chromogenic assay. (B) EVs were prepared and treated with annexin V as described in panel A. The ability of EVs to support prothrombin activation was measured by adding FVa (10 nM) and FXa (1.0 nM), followed by the substrate prothrombin (1.4 µM). Thrombin generated in the reaction mixture was measured in a chromogenic assay. (C-D) EVs, isolated from control- or FVIIa-treated HUVECs, were quantified, and an equal number of EVs were used to compare their ability to activate FX (C) and prothrombin (D). The assay conditions for measuring the rate of FX and prothrombin activation were the same as those used in panels A and B, respectively. (E-F) HUVECs were treated with FVIIa (100 nM) for 5 hours. Then, cells were washed twice to remove FVIIa and incubated with annexin V (400 nM), TF neutralizing antibody (10 µg/mL), or control IgG (10 µg/mL) for 1 hour. Intact cells were used to assess the cell surface associated-procoagulant activity in FX (E) or prothrombin (F) activation assays, as described for panels A and B. (G) HUVECs, cultured in glass coverslips, were treated with a control vehicle or FVIIa (100 nM) for 5 hours. Cells were washed and incubated with fluorescein isothiocyanate–conjugated annexin V (dilution 1:20) for 1 hour. Cells were then washed, fixed with 4% paraformaldehyde, and subjected to fluorescence microscopy (original magnification, ×40). **P < .01; ****P < .0001.
FVIIa-induced EVs exhibit higher procoagulant activity in vitro, and this is dependent on anionic phospholipids and independent of TF. (A) EVs released from HUVECs upon control vehicle or FVIIa treatment (100 nM for 24 hours) were isolated and suspended into equal amounts of a calcium-containing buffer. EVs were incubated with either annexin V (400 nM), TF neutralizing antibodies (10 µg/mL), or control IgG (10 µg/mL) for 1 h. After that, FVIIa (10 nM) was added to the EV suspension, and FX (175 nM) was added 5 minutes later. The rate of FX activation was measured in a chromogenic assay. (B) EVs were prepared and treated with annexin V as described in panel A. The ability of EVs to support prothrombin activation was measured by adding FVa (10 nM) and FXa (1.0 nM), followed by the substrate prothrombin (1.4 µM). Thrombin generated in the reaction mixture was measured in a chromogenic assay. (C-D) EVs, isolated from control- or FVIIa-treated HUVECs, were quantified, and an equal number of EVs were used to compare their ability to activate FX (C) and prothrombin (D). The assay conditions for measuring the rate of FX and prothrombin activation were the same as those used in panels A and B, respectively. (E-F) HUVECs were treated with FVIIa (100 nM) for 5 hours. Then, cells were washed twice to remove FVIIa and incubated with annexin V (400 nM), TF neutralizing antibody (10 µg/mL), or control IgG (10 µg/mL) for 1 hour. Intact cells were used to assess the cell surface associated-procoagulant activity in FX (E) or prothrombin (F) activation assays, as described for panels A and B. (G) HUVECs, cultured in glass coverslips, were treated with a control vehicle or FVIIa (100 nM) for 5 hours. Cells were washed and incubated with fluorescein isothiocyanate–conjugated annexin V (dilution 1:20) for 1 hour. Cells were then washed, fixed with 4% paraformaldehyde, and subjected to fluorescence microscopy (original magnification, ×40). **P < .01; ****P < .0001.
Next, we investigated whether the increased PS levels in FVIIa-induced EVs come from the increased externalization of PS to the outer leaflet of endothelial cells in response to FVIIa treatment. We evaluated the cell surface–associated procoagulant activity of intact endothelial cells treated with a control vehicle or FVIIa. As shown in Figure 3E-F, FVIIa treatment increased the cell-associated, PS-dependent activation of FX and prothrombin. Staining of intact HUVECs with fluorescein isothiocyanate–annexin V showed increased staining of the cell surface in FVIIa-treated cells (Figure 3G). Additional studies showed that FVIIa treatment increased the PS-dependent prothrombinase activity on the cell surface and EVs in a time-dependent manner. A significant increase was noted at 2 hours following the exposure to FVIIa, but the activity was further increased and sustained until 24 hours (supplemental Figure 8A-B). FVIIa-induced PS externalization on the endothelial cell surface appears to be a specific process. It does not reflect endothelial cell activation, as FVIIa treatment neither released von Willebrand factor (VWF) through granule exocytosis nor altered the expression of P-selectin on the cell surface (supplemental Figure 9A-C).
Experiments performed with EVs isolated from mouse brain endothelial cells further confirmed the above data. FVIIa-derived EVs from murine brain endothelial cells isolated from WT, EPCR-OX, and PAR1-R46Q mice, but not EPCR-KO and PAR1-R41Q mice, increased the rate of FX and prothrombin activation by ∼6- to 10-fold compared with EVs isolated from the corresponding cell type treated with a control vehicle (Figure 4A-B). When equal numbers of EVs from control vehicle- or FVIIa-treated WT murine brain endothelial cells were tested, FVIIa-generated EVs exhibited an enhanced FX and prothrombin activation (approximately threefold higher) compared with control EVs. In contrast, no significant differences were found between EVs generated by control vehicle or FVIIa treatment from EPCR-KO and PAR1-R41Q endothelial cells in their ability to support the activation of FX or prothrombin (supplemental Figure 10A-B). Like FVIIa-generated EVs in human endothelial cells, the increased ability of FVIIa-generated EVs from murine endothelial cells to activate FX and prothrombin was dependent on PS, as annexin V blocked the increased procoagulant activity associated with FVIIa-induced EVs. As observed with human endothelial cells, FVIIa treatment of murine brain endothelial cells increased cell surface–associated PS-dependent procoagulant activity (Figure 4C-D).

The hemostatic potential of FVIIa-derived EVs from murine brain endothelial cells isolated from various genotypes. Brain endothelial cells were isolated from WT, EPCR-KO, EPCR-OX, PAR1-R41Q, and PAR1-R46Q mice and cultured ex vivo. Confluent monolayers of endothelial cells were treated with a control vehicle or FVIIa (100 nM) for 24 hours in serum-free medium. (A-B) Equal volumes of EVs isolated from the conditioned medium were evaluated for their ability to activate FX (A) or prothrombin (B) in the presence of either control vehicle or annexin V (400 nM). (C-D) WT murine endothelial cells were treated with a control vehicle or FVIIa (100 nM) for 5 hours. The cells were washed to remove FVIIa and then treated with annexin V (400 nM) for 30 minutes. (C-D) Cell surface–associated procoagulant activity was evaluated in FX activation assay by adding FVIIa (10 nM) and FX (175 nM) (C) or prothrombin activation assay by adding FVa (10 nM), FXa (1.0 nM), and prothrombin (1.4 µM) (D). *P < .05; ***P < .001; ****P < .0001.
The hemostatic potential of FVIIa-derived EVs from murine brain endothelial cells isolated from various genotypes. Brain endothelial cells were isolated from WT, EPCR-KO, EPCR-OX, PAR1-R41Q, and PAR1-R46Q mice and cultured ex vivo. Confluent monolayers of endothelial cells were treated with a control vehicle or FVIIa (100 nM) for 24 hours in serum-free medium. (A-B) Equal volumes of EVs isolated from the conditioned medium were evaluated for their ability to activate FX (A) or prothrombin (B) in the presence of either control vehicle or annexin V (400 nM). (C-D) WT murine endothelial cells were treated with a control vehicle or FVIIa (100 nM) for 5 hours. The cells were washed to remove FVIIa and then treated with annexin V (400 nM) for 30 minutes. (C-D) Cell surface–associated procoagulant activity was evaluated in FX activation assay by adding FVIIa (10 nM) and FX (175 nM) (C) or prothrombin activation assay by adding FVa (10 nM), FXa (1.0 nM), and prothrombin (1.4 µM) (D). *P < .05; ***P < .001; ****P < .0001.
FVIIa promotes EV generation from the endothelium in vivo
In the first set of experiments, we analyzed the time course of EV release in WT mice administered with FVIIa (0.25 mg/kg of body weight). A significant increase of EVs in the circulating blood was observed at the first time point of sampling (ie, 30 minutes). The numbers of EVs in blood reached maximal levels at 2 hours and declined to basal level by 6 hours (Figure 5A). To investigate the role of EPCR and PAR1 in FVIIa-induced release of EVs in vivo, we injected saline or FVIIa (0.25 mg/kg) into WT, EPCR-KO, EPCR-OX, PAR1-R41Q, and PAR1-R46Q–mutant mice. As shown in Figure 5B, FVIIa treatment increased the generation of EVs by approximately fourfold in WT, EPCR-OX, and PAR1-R46Q–mutant mice, but not in EPCR-KO and PAR1-R41Q mice.
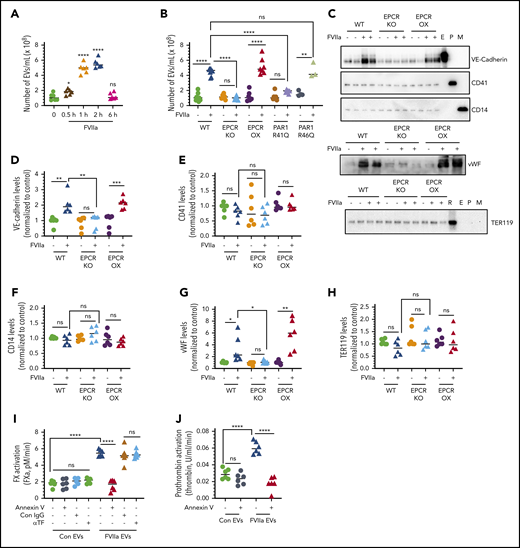
FVIIa treatment induces the release of procoagulant EVs from the endothelium in vivo via EPCR and PAR1 activation–dependent mechanism. (A) WT mice were administered with FVIIa (0.25 mg/kg body weight) via the tail vein. At varying times following FVIIa administration, mice were killed, blood was collected, and EVs were isolated from the plasma and counted by NTA NanoSight. (B) WT, EPCR-KO, EPCR-OX, PAR1-R41Q, and PAR1-R46Q mice were injected with saline or FVIIa (0.25 mg/kg) via the tail vein. After 2 hours, blood was collected, and the number of EVs in the blood was quantified. (C) EVs isolated from the blood of WT, EPCR-KO, or EPCR-OX mice treated with saline or FVIIa (0.25 mg/kg) for 2 hours were subjected to immunoblot analysis to probe for VE-cadherin (endothelial cell marker), CD41 (platelet marker), CD14 (monocyte marker), TER119 (RBC marker), or VWF. Lysates of murine brain endothelial cells (E), platelets (P), peripheral blood mononuclear cells (M), and RBC lysate (R) were used as positive controls and to attest to the specificity of antibodies. (D-H) Band intensities of immunoblots were quantified by densitometric analysis. (I-J) The procoagulant activity of EVs generated in vivo. EVs, isolated from WT mice treated with saline or FVIIa as described in panel B, were assayed for their ability to support the activation of FX (I) or prothrombin (J). *P < .05; **P < .01; ***P < .001; ****P < .0001.
FVIIa treatment induces the release of procoagulant EVs from the endothelium in vivo via EPCR and PAR1 activation–dependent mechanism. (A) WT mice were administered with FVIIa (0.25 mg/kg body weight) via the tail vein. At varying times following FVIIa administration, mice were killed, blood was collected, and EVs were isolated from the plasma and counted by NTA NanoSight. (B) WT, EPCR-KO, EPCR-OX, PAR1-R41Q, and PAR1-R46Q mice were injected with saline or FVIIa (0.25 mg/kg) via the tail vein. After 2 hours, blood was collected, and the number of EVs in the blood was quantified. (C) EVs isolated from the blood of WT, EPCR-KO, or EPCR-OX mice treated with saline or FVIIa (0.25 mg/kg) for 2 hours were subjected to immunoblot analysis to probe for VE-cadherin (endothelial cell marker), CD41 (platelet marker), CD14 (monocyte marker), TER119 (RBC marker), or VWF. Lysates of murine brain endothelial cells (E), platelets (P), peripheral blood mononuclear cells (M), and RBC lysate (R) were used as positive controls and to attest to the specificity of antibodies. (D-H) Band intensities of immunoblots were quantified by densitometric analysis. (I-J) The procoagulant activity of EVs generated in vivo. EVs, isolated from WT mice treated with saline or FVIIa as described in panel B, were assayed for their ability to support the activation of FX (I) or prothrombin (J). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Next, we investigated the cellular source of EVs generated by FVIIa in the circulation. EVs isolated from WT, EPCR-KO, and EPCR-OX mice challenged with saline or FVIIa were subjected to immunoblot analysis and probed with endothelial cell–, platelet-, monocyte-, and RBC-specific markers with appropriate positive controls. As shown in Figure 5C, there was a marked increase in an endothelial cell–specific marker, VE-cadherin, in EVs isolated from WT and EPCR-OX mice challenged with FVIIa over the mice administered with saline. VE-cadherin levels were not increased significantly in EVs isolated from EPCR-KO mice challenged with FVIIa (Figure 5C-D). FVIIa-released EVs from WT and EPCR-OX mice were also enriched with VWF, which was absent in EVs released from EPCR-KO mice (Figure 5C-G). We found no significant increase in the platelet-specific marker CD41, the monocyte-specific marker CD14, or the RBC-specific marker TER119 in EVs isolated from all 3 genotypes following FVIIa administration (Figure 5C,E-F,H). These data indicate that vascular endothelial cells are the cellular source of EVs released into circulating blood following FVIIa administration.
Similar to that observed with EVs released from cultured endothelial cells in response to FVIIa, EVs isolated from blood following FVIIa administration to WT mice supported the activation of FX and prothrombin at a threefold higher rate than EVs isolated from saline-administered WT mice (equal number of EVs) (Figure 5I-J). Annexin V treatment completely abolished the enhanced FX and prothrombin activation by FVIIa-generated EVs, whereas anti-TF antibodies had no effect on FX activation (Figure 5I-J). No significant differences were observed between control- and FVIIa-generated EVs from EPCR-KO and PAR1-R41Q mice in their ability to support the activation of FX or prothrombin (supplemental Figure 11A-D).
FVIIa-induced EVs promote hemostasis in vivo
To investigate the potential hemostatic effect of FVIIa-induced endothelial EVs, we administered equal numbers of control- or FVIIa-derived EVs (1 × 109 EVs per mouse) to platelet-depleted WT mice, and bleeding was induced by the saphenous vein incision. The administration of CD42b mAb to WT mice depleted the platelet count by >90% (platelet count was 1.08 ± 0.03 × 106/µL before the administration of the antibody and 0.09 ± 0.01 × 106/µL after the administration of the antibody; Figure 6A). As shown in Figure 6B, the administration of FVIIa-generated EVs to platelet-depleted mice significantly reduced the blood loss following the injury. Administration of the same number of control EVs had no significant effect on blood loss in platelet-depleted mice. The measurement of TAT levels in the blood collected at the injury site showed a significant increase in TAT levels in platelet-depleted mice injected with FVIIa-derived EVs, but not control EVs (Figure 6C).
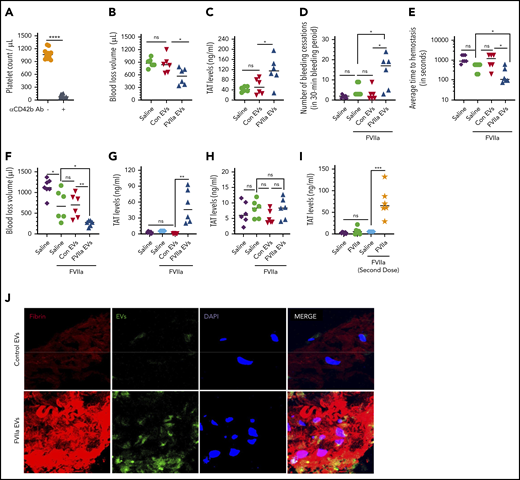
The hemostatic potential of FVIIa-induced EVs. (A) Platelet depletion upon the administration of antiplatelet antibody (αCD42b Ab). WT mice were injected with CD42b antibodies (1 mg/kg) via the tail vein. Platelet counts in the blood were measured before the administration of the antibody and 5 hours following antibody administration. (B) Infusion of FVIIa-induced EVs to platelet-depleted mice reduces blood loss following saphenous vein injury. WT mice were injected with CD42b antibodies (1 mg/kg) via the tail vein to deplete platelets. Five hours following the administration of platelet-depleting antibodies, mice were injected with equal numbers of EVs (1 × 109/mouse) generated from endothelial cells treated with a control vehicle, FVIIa (100 nM), or saline. Immediately following the administration of EVs, mice were subjected to the saphenous vein incision, and the blood coming from the injury site was collected on filter paper. Hemoglobin was extracted from the filter paper, and values were extrapolated to the blood volume using a standard curve derived with known volumes of blood. (C) FVIIa-induced EVs support the generation of thrombin at the wound site in platelet-depleted mice. WT mice were depleted of platelets, administered with control or FVIIa-generated endothelial EVs, and subjected to saphenous vein incision as described for panel B. Blood coming from the wound site was collected periodically (up to 15 minutes; if a clot was formed, it was dislodged) directly into citrate anticoagulant, and thrombin generation was measured as the amount of TAT complexes formed in the blood. (D-H) FVIIa-generated EVs support FVIIa-induced hemostasis in hemophilia mice. FVIII−/− mice were injected with saline or EVs derived from endothelial cells treated with a control vehicle or FVIIa (1.5 × 109/mouse). Immediately following EVs, mice were administered with saline or FVIIa (0.25 mg/kg), a dose well below the threshold of FVIIa needed to correct the bleeding in itself, and subjected to the saphenous vein incision. The number of hemostatic plugs formed in a 30-minute interval was recorded (D), and the average time to achieve hemostasis (E) and blood loss (F) were determined. (G) TAT levels in the blood collected at the wound site. (H) TAT levels in the blood collected via the submandibular vein. (I) Effect of endogenously generated EVs on thrombin generation at the wound site. FVIII−/− mice were treated with a control vehicle control or rFVIIa (0.25 mg/kg) to generate EVs endogenously. After 2 hours, mice were subjected to saphenous vein incision with or without giving a second dose of rFVIIa (0.25 mg/kg) immediately prior to saphenous vein incision. Blood leaking from the wound site was collected, and levels of TAT complexes were determined as described in panel G. (J) Recruitment of FVIIa-generated EVs to the wound site. Equal numbers (1.5 × 109 EVs) of PKH67-labeled (green fluorescence) EVs derived from murine b.END3 endothelial cells treated with a control vehicle or FVIIa were injected into FVIII−/− mice via the tail vein, followed by FVIIa (0.25 mg/kg) administration. Mice were subjected to saphenous vein incision. After 5 minutes, the injured saphenous vein was excised, fixed, and processed for sectioning. Sections were stained for fibrin using antibodies against murine fibrinogen (red), and nuclei were stained with DAPI (blue) and analyzed by confocal microscopy (original magnification ×63 with 2.4× zoom) . *P < .05; **P < .01; ***P < .001; ****P < .0001.
The hemostatic potential of FVIIa-induced EVs. (A) Platelet depletion upon the administration of antiplatelet antibody (αCD42b Ab). WT mice were injected with CD42b antibodies (1 mg/kg) via the tail vein. Platelet counts in the blood were measured before the administration of the antibody and 5 hours following antibody administration. (B) Infusion of FVIIa-induced EVs to platelet-depleted mice reduces blood loss following saphenous vein injury. WT mice were injected with CD42b antibodies (1 mg/kg) via the tail vein to deplete platelets. Five hours following the administration of platelet-depleting antibodies, mice were injected with equal numbers of EVs (1 × 109/mouse) generated from endothelial cells treated with a control vehicle, FVIIa (100 nM), or saline. Immediately following the administration of EVs, mice were subjected to the saphenous vein incision, and the blood coming from the injury site was collected on filter paper. Hemoglobin was extracted from the filter paper, and values were extrapolated to the blood volume using a standard curve derived with known volumes of blood. (C) FVIIa-induced EVs support the generation of thrombin at the wound site in platelet-depleted mice. WT mice were depleted of platelets, administered with control or FVIIa-generated endothelial EVs, and subjected to saphenous vein incision as described for panel B. Blood coming from the wound site was collected periodically (up to 15 minutes; if a clot was formed, it was dislodged) directly into citrate anticoagulant, and thrombin generation was measured as the amount of TAT complexes formed in the blood. (D-H) FVIIa-generated EVs support FVIIa-induced hemostasis in hemophilia mice. FVIII−/− mice were injected with saline or EVs derived from endothelial cells treated with a control vehicle or FVIIa (1.5 × 109/mouse). Immediately following EVs, mice were administered with saline or FVIIa (0.25 mg/kg), a dose well below the threshold of FVIIa needed to correct the bleeding in itself, and subjected to the saphenous vein incision. The number of hemostatic plugs formed in a 30-minute interval was recorded (D), and the average time to achieve hemostasis (E) and blood loss (F) were determined. (G) TAT levels in the blood collected at the wound site. (H) TAT levels in the blood collected via the submandibular vein. (I) Effect of endogenously generated EVs on thrombin generation at the wound site. FVIII−/− mice were treated with a control vehicle control or rFVIIa (0.25 mg/kg) to generate EVs endogenously. After 2 hours, mice were subjected to saphenous vein incision with or without giving a second dose of rFVIIa (0.25 mg/kg) immediately prior to saphenous vein incision. Blood leaking from the wound site was collected, and levels of TAT complexes were determined as described in panel G. (J) Recruitment of FVIIa-generated EVs to the wound site. Equal numbers (1.5 × 109 EVs) of PKH67-labeled (green fluorescence) EVs derived from murine b.END3 endothelial cells treated with a control vehicle or FVIIa were injected into FVIII−/− mice via the tail vein, followed by FVIIa (0.25 mg/kg) administration. Mice were subjected to saphenous vein incision. After 5 minutes, the injured saphenous vein was excised, fixed, and processed for sectioning. Sections were stained for fibrin using antibodies against murine fibrinogen (red), and nuclei were stained with DAPI (blue) and analyzed by confocal microscopy (original magnification ×63 with 2.4× zoom) . *P < .05; **P < .01; ***P < .001; ****P < .0001.
Next, we investigated the effect of FVIIa-induced EVs in correcting the bleeding in FVIII−/− mice. FVIII−/− mice were administered with control EVs or FVIIa-induced EVs isolated from HUVECs via the tail vein. Immediately following EV administration, FVIII−/− mice were injected with a low dose of FVIIa (0.25 mg/kg), which would not be enough to correct the severe bleeding in FVIII−/− mice following the injury. As shown in Figure 6D, administration of 0.25 mg/kg FVIIa alone had no significant effect on bleeding times. However, the administration of FVIIa-induced EEVs increased the number of hemostatic plugs formed in the 30-minute bleeding period. The average time to achieve hemostasis was decreased from 1243 ± 201 seconds to 224 ± 86 seconds (Figure 6E). Administration of the same number of control EEVs had no significant effect on the bleeding. Similarly, total blood loss was reduced significantly upon the administration of FVIIa-derived EEVs as compared with either control EEVs or saline (Figure 6F). Significant thrombin generation was observed at the wound site in mice administered with FVIIa-generated EEVs, but not control EEVs (Figure 6G). Consistent with the observation that FVIIa-derived EEVs promote clotting, plasma fibrinogen levels were significantly reduced in mice administered with FVIIa-generated EEVs, but not control EEVs (supplemental Figure 12). Measurement of TAT levels in blood drawn via a submandibular vein in the above groups of mice showed no significant differences in TAT levels (Figure 6H), indicating FVIIa-induced EVs do not promote activation of systemic coagulation.
In additional experiments to investigate the hemostatic effect of endogenously derived EEVs, we first treated FVIII−/− mice with saline or FVIIa (0.25 mg/kg). After 2 hours (by which time the levels of FVIIa-released EEVs in the circulation would reach the peak), mice were subjected to saphenous vein incision. Since FVIIa administered to mice would have been cleared completely from the circulation in 2 hours,50 a group of mice was also injected again with the second dose of FVIIa (0.25 mg/kg) immediately prior to saphenous vein incision. In the absence of circulating FVIIa at the time of injury, the generation of endogenous EEVs by FVIIa pretreatment failed to generate thrombin at the wound site (second bar in Figure 6I). However, FVIIa-generated endogenous EEVs markedly enhanced thrombin generation at the wound site if FVIIa was also available in the circulation along with EEVs at the time of injury (fourth bar in Figure 6I).
To determine whether the enhanced hemostatic effect of FVIIa-derived EEVs stems from their recruitment to the wound site selectively, we administered equal numbers (1.5 × 109 EVs) of PKH67-labeled control vehicle- or FVIIa-generated EVs from murine endothelial cells to FVIII−/− mice. Following EV administration, mice were injected with FVIIa (0.25 mg/kg) and immediately subjected to saphenous vein incision. After 5 minutes, the wound site was excised, fixed, and sectioned, and the sections were stained for fibrin and subjected to confocal microscopy. Administration of FVIIa-derived EEVs generated more fibrin at the wound site than administration of control EEVs. More importantly, FVIIa-derived EEVs and not control EEVs were found in the fibrin clot at the wound site (Figure 6J). Furthermore, blood collected from the incision site of mice injected with PKH67-labeled FVIIa-derived EEVs showed markedly higher fluorescence than blood collected from mice administered with an equal number of PKH67-labeled control EEVs (supplemental Figure 13A). The fluorescence intensity of blood collected from the submandibular vein was similar in mice injected with either labeled control or FVIIa-derived EEVs (supplemental Figure 13B). Overall, these data indicate that FVIIa-derived EEVs were selectively recruited to the wound site.
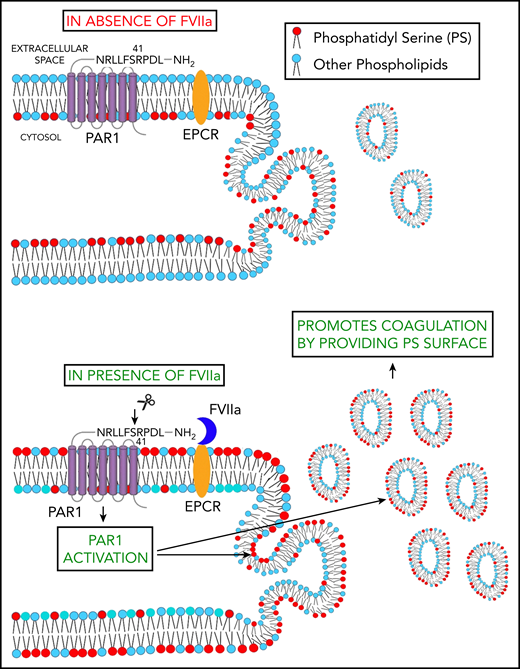
Schematic representation of FVIIa-induced EV release. FVIIa, upon binding to EPCR on endothelial cells, cleaves PAR1 at the canonical R41 cleavage site. This results in the externalization of PS to the outer leaflet of the plasma membrane. FVIIa activation of PAR1 also leads to the budding of EVs from endothelial cells. Thus, FVIIa-induced EVs are enriched with PS and capable of supporting FVIIa-induced hemostasis in hemophilia patients and patients with platelet disorders.
Schematic representation of FVIIa-induced EV release. FVIIa, upon binding to EPCR on endothelial cells, cleaves PAR1 at the canonical R41 cleavage site. This results in the externalization of PS to the outer leaflet of the plasma membrane. FVIIa activation of PAR1 also leads to the budding of EVs from endothelial cells. Thus, FVIIa-induced EVs are enriched with PS and capable of supporting FVIIa-induced hemostasis in hemophilia patients and patients with platelet disorders.
Discussion
A series of studies from our laboratory showed that FVIIa binding to EPCR on the endothelium leads to the activation of PAR1-mediated biased cell signaling that transduces vascular barrier protective and anti-inflammatory effects.14-17 EPCR, having a very short cytoplasmic tail (3 amino acids long), is incapable of inducing intracellular signaling of its own upon binding FVIIa but supports FVIIa activation of PAR1-mediated signaling.13,51 Until now, it was unknown whether EPCR-FVIIa–induced cell signaling also impacts the hemostatic effect of FVIIa. Here, we provide compelling evidence, for the first time, that FVIIa induces the release of EVs from endothelial cells via activation of EPCR-PAR1–mediated cell signaling. FVIIa-released EEVs exhibit higher procoagulant activity compared with EEVs released in basal conditions. In vivo studies demonstrate that FVIIa-induced EEVs contribute to the hemostatic effect by supporting the generation of thrombin at the wound site in platelet-depleted and hemophilia mouse models. Our current findings not only add to mechanistic insights into the hemostatic effect of rFVIIa in therapeutic conditions but also raise the possibility that FVIIa influences other biological processes via intercellular communication through EEVs
The evidence that FVIIa induces the release of EVs from endothelial cells through the EPCR-PAR1 axis comes from both in vitro cell model systems and in vivo murine transgenic animal model systems. Knockdown of either EPCR or PAR1 completely abrogated the FVIIa-induced release of EVs in human and murine endothelial cells. In vivo studies showed that the administration of FVIIa to WT, EPCR-OX, and PAR1-R46Q mice, but not EPCR-KO and PAR1-R41Q mice, increased levels of circulating EEVs. The present data showing that PAR1 mutation at the canonical activation site R41 blocks FVIIa-induced EEV release are consistent with our recent findings that EPCR-FVIIa–induced PAR1 biased signaling is mediated through cleavage of PAR1 at the R41 site.17 The differences in the time course of EVs reaching a peak in response to FVIIa in vitro and in vivo may reflect potential differences in the clearance of EVs in vitro and in vivo. The balance between secretion and clearance determines the concentration of EVs in the blood in vivo, and multiple mechanisms could play a role in the clearance of EVs in vivo.52,53
A vast majority of circulating plasma EVs are derived from platelets and erythrocytes.24,26 Although endothelial cells constitutively secrete a low number of EVs under physiological conditions, various diseases associated with endothelial injury or dysfunction have been found to elevate the release of EEVs into plasma.25,30,54 Various proinflammatory cytokines, lipopolysaccharide, and thrombin were shown to release EEVs both in vitro and in vivo.55 Although increased levels of EEVs in circulation, in general, are considered to be detrimental, as they impair the vascular function by being prothrombotic and proinflammatory, they could also display anticoagulant, anti-inflammatory, and cytoprotective effects.56 There is no solid evidence that EEVs secreted from healthy subjects promote coagulation. Only EEVs derived from the activated endothelium were found to be procoagulant or prothrombotic, as these EEVs contain TF on their surface.30,31,34 FVIIa-induced EEVs, unless they come from dysfunctional endothelium expressing TF, will not contain TF (supplemental Figure 14). Therefore, it is unlikely that FVIIa-derived EEVs induce systemic activation of the coagulation or vascular thrombosis.
The role of EEVs in hemostasis is unknown. The data presented in the current paper show that although EEVs generated in normal physiological conditions are unlikely to contribute to hemostasis, EEVs generated in response to FVIIa treatment can support hemostasis at the site of injury. Our data show that the administration of FVIIa-generated EEVs supports thrombin generation at the wound site and reduces blood loss in platelet-depleted or hemophilia mice subjected to saphenous vein incision. It is pertinent to note here not only that FVIIa treatment increased the number of EVs released from endothelial cells but also that these EVs also appear to contain higher PS levels on their surface, as they support FVIIa-catalyzed activation of factor X and FXa-catalyzed activation of prothrombin more readily compared with EEVs generated in basal conditions, which are known to contain PS.24,57,58 The increased levels of PS in FVIIa-generated EEVs are likely to come from the externalization of PS on the endothelial cell surface following FVIIa treatment (Figure 7). We are not aware of any prior report that showed FVIIa induces the externalization of PS. At present, the potential mechanisms by which FVIIa promotes PS externalization on endothelial cells is unknown. However, FVIIa-induced PS externalization was not due to either endothelial cell activation or apoptosis, as FVIIa induces neither endothelial cell activation nor apoptosis. Earlier studies showed that cells could externalize PS without undergoing apoptosis.59-61
Earlier studies reported that APC induces the release of EVs from endothelial cells in the EPCR- and PAR1-dependent mechanism, and the APC-induced EEVs express anticoagulant activity62 and induce cytoprotective and anti-inflammatory effects.63 These studies also showed that APC-induced EEVs retained a large amount of APC (10 to 40 nM), and this APC was responsible for the anticoagulant activity and cytoprotective effects observed with APC-derived EEVs.62,63 We found no evidence in our current studies that FVIIa-induced EEVs display such a mechanism. We were unable to detect measurable FVIIa in EEVs either by immune blot analysis or FVIIa activity assay. It is likely that a small fraction of FVIIa might have associated with EPCR embedded in EEVs, but most of it could have been dissociated during the isolation of EEVs. It is unlikely that FVIIa-induced EEVs in vivo retain significant amounts of FVIIa in the circulation, as our earlier studies showed that FVIIa administered to mice was rapidly cleared from the circulation.50 FVIIa was barely detectable in blood at 2 hours following its administration,50 whereas FVIIa-induced EEV levels peaked at 2 hours following FVIIa treatment.
At present, it is not entirely clear how FVIIa-induced EEVs provide the hemostatic effect. It is important to reiterate here that FVIIa-induced EEVs do not generate thrombin in circulation. EEV-induced thrombin generation is limited to the wound site. It is possible that EEVs act as mini-activated platelets at the wound site. It is also conceivable that FVIIa-induced EEVs can fuse with platelets and leukocytes in the circulation or at the wound site and promote their hemostatic activity. It is believed by some investigators that EVs in the blood of healthy individuals contain TF but are unable to generate thrombin, as PS levels in EVs may be below the optimal levels to support TF activity.58 It is possible that FVIIa-induced EEVs can fuse with such TF-positive EVs and potentiate their ability to generate thrombin at the site of injury. A careful and detailed study is required to investigate the above possibilities.
In our earlier studies, we showed that FVIIa induces EPCR-dependent PAR1-mediated cell signaling and that FVIIa-EPCR-PAR1 signaling promotes vascular barrier protective and anti-inflammatory effects in vivo.14-17 It is possible that FVIIa-induced EEVs may be playing a role in mediating FVIIa-induced barrier protective and anti-inflammatory effects. In general, the interaction between EEVs and native endothelial cells triggers proinflammatory responses.64-67 However, EEVs were also shown to exert beneficial effects, such as promoting EC survival.55,68 EEVs were shown to promote anti-inflammatory effects in vitro and in vivo in recipient endothelial cells by transfer microRNA-222.69 EEVs containing anti-inflammatory microRNA were shown to suppress monocyte activation.70 Although APC-generated EEVs were shown to induce cytoprotective and anti-inflammatory effects, these protective effects appeared to be mediated by the action of APC associated with EEVs rather than EEVs per se.63 Our preliminary studies show that FVIIa-generated EEVs induce endothelial barrier protective and anti-inflammatory effects in endothelial cells and anti-inflammatory effects in monocytes (data not shown). However, detailed studies are needed to confirm these data and establish the relevance of FVIIa-induced EEVs in regulating inflammation and vascular leakage.
The observation that FVIIa treatment generates EEVs may have a relevance to clinical settings. First, it adds mechanistic insights into how rFVIIa therapy could provide hemostatic activity in treating various bleeding disorders. rFVIIa was used effectively in treating bleeding disorders in patients with congenital and acquired platelet disorders.71,72 Our recent studies indicated that ∼30% of thrombin generated at the injury site in hemophilia mice following FVIIa administration comes from a platelet-independent mechanism.73 It is possible that FVIIa-induced EEVs may augment thrombin generation at the wound site by acting as “mini platelets” in case of acute platelet depletion. This could explain how FVIIa provides the hemostatic effect in patients with severe platelet disorders. rFVIIa is also used “off label” to treat life-threatening severe bleeding in various clinical scenarios, such as sepsis, trauma, and surgery.4,74-76 Although rFVIIa treatment was proven to be safe in most clinical situations, there is still a 1% to 10% risk of developing thromboembolic events with off-label use, particularly in patients with cardiovascular diseases.4,77 Patients with cardiovascular risk factors such as hypertension, diabetes, dyslipidemia, and smoking, as well as those with acute coronary syndromes, are known to have elevated levels of TF, including aberrant expression of TF on endothelial cells.78 If endothelial cells express TF, then it is likely to be incorporated into EEVs generated by FVIIa. The levels of TF in FVIIa-induced EEVs could determine the risk of developing thromboembolic complications upon rFVIIa treatment in these groups of patients. A caveat in our study is that it does not reveal the relative contribution of FVIIa-generated EEVs or FVIIa-induced externalization of PS on intact endothelium to the hemostatic effect in correcting bleeding disorders, as it is not feasible to target PS selectively on EVs, endothelial cells, and platelets. Since FVIIa treatment does not induce thrombin generation in circulating blood, it is unlikely that either FVIIa-generated EVs or PS externalization on the endothelium drives systemic coagulation. The release of EVs from the endothelium by FVIIa and the recruitment of FVIIa-released EEVs to the wound site may contribute substantially to the hemostatic effect of pharmacological concentration of FVIIa. However, we cannot preclude the role of FVIIa-perturbed endothelium at the wound site contributing to thrombin generation.
Overall, our current findings demonstrate that FVIIa induces the release of EVs from endothelial cells via EPCR-PAR1–mediated biased signaling (Figure 7). Our studies also provide a proof of concept that EEVs generated by FVIIa treatment can contribute to hemostasis. This novel observation adds new insights into the mechanism of FVIIa’s hemostatic activity in FVIIa therapy and links FVIIa signaling to the hemostatic process. Our data may impact future FVIIa therapy modalities.
The authors will share all methodologies and original data sets to qualified investigators upon specific request to the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL107483 and HL124055 (L.V.M.R.), UM1 HL120877 (C.T.E.), and HL052246 and HL142975 (J.H.G.); endowment funds from The Dr and Mrs James Vaughn Professorship in Biomedical Research (L.V.M.R.), and a Judith Graham Pool Fellowship award from the National Hemophilia Foundation (K.D.).
Authorship
Contribution: K.D. performed a majority of studies described in the paper, analyzed the data, and wrote an initial draft of the manuscript; S.K. performed animal studies and immunohistochemistry; S.A.A. and V.K. performed a few in vitro studies; U.R.P. contributed to the study design, provided technical expertise in performing the study, and participated in data analysis and manuscript preparation; C.T.E. provided breeding pairs of EPCR-KO and EPCR-OX mice and EPCR antibodies; J.H.G. provided the breeding pairs of PAR1 mutant mice; L.V.M.R. conceived and designed the research, analyzed data, and wrote the manuscript; and all authors contributed to the preparation of the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: L. Vijaya Mohan Rao, Department of Cellular and Molecular Biology, The University of Texas Health Science Center at Tyler, Tyler, TX 75708-3154; e-mail: vijay.rao@uthct.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal