

TO THE EDITOR:
The introduction of ibrutinib (along with other Bruton tyrosine kinase inhibitors such as the recently approved acalabrutinib) and venetoclax has led to a change of paradigm in treatment of chronic lymphocytic leukemia (CLL).1,2 The logical next step is targeting Bruton tyrosine kinase of the B-cell receptor pathway by ibrutinib combined with targeting B-cell lymphoma 2 of the antiapoptotic pathway by venetoclax.3,4 We here demonstrate high rates of complete remission (CR) with increasing rates of undetectable minimal residual disease (MRD; uMRD) over time by ibrutinib plus venetoclax in relapsed or refractory (R/R) CLL.5 For R/R CLL, different combination approaches are currently being tested in clinical trials.6,7 Venetoclax-rituximab in the R/R CLL setting, although offering a time-limited treatment, does not provide high rates of CR.2 Thus, there is an urgent need for chemotherapy-free regimens that result in deep responses, preventing the need for prolonged maintenance. Preclinical data support combining ibrutinib and venetoclax, demonstrating synergy in cell lines,8-11 in primary CLL cells and a CLL mouse model,11 whereas recently, efficacy of venetoclax plus ibrutinib in first-line and R/R CLL has been reported.3,4 uMRD correlates with longer remissions in CLL upon chemoimmunotherapy as well as targeted therapy.7,12 Consequently, the aim of the current trial is (1) to evaluate whether combination treatment with venetoclax plus ibrutinib in patients with R/R CLL can lead to uMRD with long-lasting remissions and (2) to test an MRD-guided treatment approach for stopping and reinitiating targeted agents in R/R CLL.
In the VISION phase 2 study (clinicaltrials.gov #NCT03226301), patients diagnosed with R/R CLL after 1 or more lines of prior chemoimmunotherapy with indication for treatment according to International Workshop on CLL (IWCLL) 2008 criteria13 were eligible. Patients were eligible independent of TP53 aberrations and comorbidities as long as they had a creatinine clearance ≥30 mL/min. All patients received ibrutinib monotherapy (420 mg daily) for 2 cycles. Venetoclax ramp-up according to the approved label was initiated during cycle 3, reaching the full dosage of 400 mg daily from the start of cycle 4 with continued combination treatment of a further 12 cycles, totaling 15 cycles (28-day cycles). Patients reaching at least partial remission (PR) and uMRD in blood and bone marrow (BM) at cycle 15 are randomized between maintenance ibrutinib or observation; combination treatment is reinitiated for patients becoming MRD positive (MRD+) during observation. Patients MRD+ at cycle 15 remain on ibrutinib maintenance therapy. Patients becoming MRD+ on ibrutinib remain on ibrutinib; if there is progressive disease, these patients will go off protocol. Reinitiating treatment due to MRD+ is not considered progression if remission is maintained or achieved (please see the published protocol for further details14 ). We report the preplanned analysis for the first 51 eligible patients reaching cycle 15.
Enrollment was completed with 230 patients enrolled between July 2017 and January 2019. The trial population is representative of previously published R/R CLL trials (supplemental Table 1, available on the Blood Web site), with the following characteristics: median age of 67 years (range, 40-83 years); 71% of patients were male; 65% had a World Health Organization (WHO) performance status of 0; 84% were Binet stage B/C; 71% received prior standard chemoimmunotherapy; 18% had TP53 aberrations (deletion 17p and/or TP53 mutation, centrally assessed); and 57% had immunoglobulin heavy chain–unmutated status (centrally assessed).2,15 Of the 51 patients in this preplanned interim analysis including data until cycle 15, 49 patients completed the first 2 cycles of ibrutinib monotherapy and 43 (84%) completed all 15 cycles (supplemental Results; supplemental Figure 1). CR was achieved by 27 patients (53%); the remaining 15 patients (29%) with full IWCLL assessment achieved PR (Figure 1A).
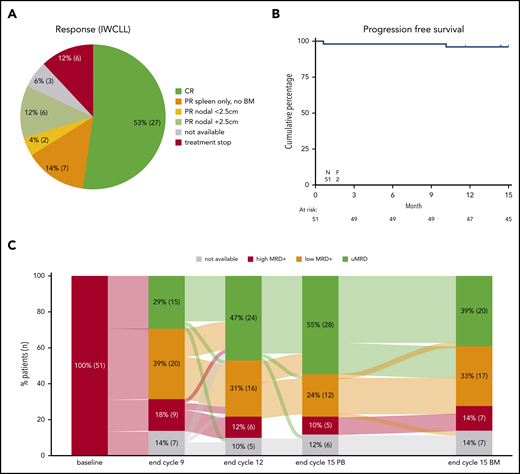
Responses and outcome. (A) Response according to IWCLL criteria for the first 51 patients. “PR spleen only, no BM” indicate that a spleen diameter >13 cm was the only criteria for not achieving CR; 2 patients without BM data available are also included here. PR nodal <2.5 cm indicates that no lymph nodes were >2.5 cm in largest diameter; PR nodal +2.5 cm indicates that lymph nodes >2.5 cm in diameter were present. For the 6 patients stopping treatment, no response data were available; this includes the 2 fatalities. The 3 patients with unavailable response assessment represent 2 patients with clinical CR and 1 patient with clinical CR incomplete recovery due to thrombocytopenia. (B) Progression-free survival. The 2 fatal events are depicted; no progressions were reported. (C) Sankey plot for minimal residual response (MRD) of the first 51 eligible patients completing cycle 15. Kinetics is shown on PB and BM aspirates. uMRD indicates undetectable MRD <10−4 by flow cytometry; low MRD+ indicates MRD levels between 10−2 and 10−4; high MRD+ indicates MRD levels ≥10−2. To the right, MRD response in BM is shown for comparison with MRD response in PB at cycle 15; end of cycle 9 and end of cycle 12 samples are PB.
Responses and outcome. (A) Response according to IWCLL criteria for the first 51 patients. “PR spleen only, no BM” indicate that a spleen diameter >13 cm was the only criteria for not achieving CR; 2 patients without BM data available are also included here. PR nodal <2.5 cm indicates that no lymph nodes were >2.5 cm in largest diameter; PR nodal +2.5 cm indicates that lymph nodes >2.5 cm in diameter were present. For the 6 patients stopping treatment, no response data were available; this includes the 2 fatalities. The 3 patients with unavailable response assessment represent 2 patients with clinical CR and 1 patient with clinical CR incomplete recovery due to thrombocytopenia. (B) Progression-free survival. The 2 fatal events are depicted; no progressions were reported. (C) Sankey plot for minimal residual response (MRD) of the first 51 eligible patients completing cycle 15. Kinetics is shown on PB and BM aspirates. uMRD indicates undetectable MRD <10−4 by flow cytometry; low MRD+ indicates MRD levels between 10−2 and 10−4; high MRD+ indicates MRD levels ≥10−2. To the right, MRD response in BM is shown for comparison with MRD response in PB at cycle 15; end of cycle 9 and end of cycle 12 samples are PB.
The cumulative incidence of serious adverse events (SAEs) during the first 15 cycles was 57% (Figure 2A). The distribution of types of AEs by highest grade is summarized in supplemental Table 2. One patient died due to unknown cause during ibrutinib monotherapy and 1 patient died subsequent to lung cancer during ibrutinib and venetoclax combination therapy. For AEs of special interest (grade ≥2), 9 patients (18%) were reported with atrial fibrillation (all grade 2, except 1 grade 3) and 9 (18%) with bleeding (all grade 2, except 1 grade 3; Figure 2B), while 34 patients (67%) were reported with infections (1 grade 4, 19 grade 3, and 14 grade 2). The most frequent site of infections was the airway followed by the urinary tract and skin; cumulative incidences of the different sites of infections are detailed in Figure 2C.
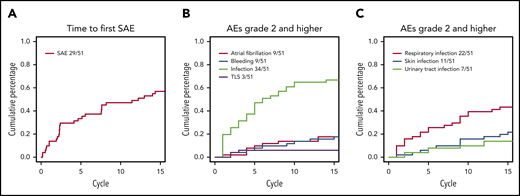
Cumulative incidence of AEs over time. (A) SAEs over time. (B) Selected AEs with CTCAE grade 2 or higher. (C) Selected categories of infections. Only first event for each type of infection for each patient included. TLS, tumor lysis syndrome.
Cumulative incidence of AEs over time. (A) SAEs over time. (B) Selected AEs with CTCAE grade 2 or higher. (C) Selected categories of infections. Only first event for each type of infection for each patient included. TLS, tumor lysis syndrome.
The depth of peripheral blood (PB) response continued to improve during treatment, with 28 (55%) reaching uMRD at cycle 15; 20 (39%) also obtained BM uMRD (intention-to-treat analysis, 10−4 level by flow cytometry16 ) (Figure 1C). The uMRD rate in PB is similar to the previously published venetoclax plus rituximab combination for R/R CLL, whereas the CR rate doubled,2 in accordance with previous early data for ibrutinib plus venetoclax.3,4 From the 43 patients (who completed all 15 cycles of therapy), 14 patients with uMRD in both BM and PB after 15 cycles of treatment have been randomized 2:1 between observation and ibrutinib maintenance according to the protocol.
Treatment with ibrutinib and venetoclax in the setting of R/R CLL demonstrates a favorable benefit-risk profile consistent with the known safety profile for the individual drugs, in line with recently reported first-line and R/R CLL results.3,4 The AE profile reported in the current study reflects previously reported data, although the incidence of atrial fibrillation at 20% is higher than reported in previous studies with monotherapy ibrutinib17 and combination studies of venetoclax plus ibrutinib.3,4 Although atrial fibrillation appeared at a steady rate during the 15 cycles of induction therapy, infections appeared at a higher frequency during the first 3 cycles of treatment, in accordance with previous reports on decreasing rates of infections over time on targeted therapy for CLL.18,19
The response rates for the combination of ibrutinib and venetoclax in the R/R CLL setting (53% CR and 30% PR) are similar to those reported by the UK group, whereas still lower than the 88% CR rate reported for this combination in first-line treatment.3,4 For venetoclax plus rituximab in R/R CLL, a much lower CR rate of 8.2% was reported, whereas comparable uMRD rates were seen.2,7 This reflects different efficacy of different treatment regimens in targeting different compartments of the disease, that is, lymph nodes, spleen, BM, and PB.20 Thus, assessment of MRD dynamics and response evaluation should take into account the type of targeted treatment regimen applied and be analyzed in the context of clinical response and molecular characterization of the disease.12
This interim analysis confirms prior results indicating efficacy and good tolerability of fully oral targeted therapy in R/R CLL.3-5 Inspired by management of chronic myelogenous leukemia,21 we here test whether combination treatment with ibrutinib plus venetoclax can be safely stopped in the R/R CLL setting for patients with uMRD disease in both BM and PB. The primary analysis of the study, testing (1) whether patients with uMRD in BM and PB at cycle 15 can remain progression free without further treatment and (2) whether reinitiating ibrutinib plus venetoclax for patients becoming MRD+ during observation can prevent clinical progression, is expected by the end of 2021.
Parts of the data presented here have been published as an earlier interim analysis (only 6 cycles of follow-up)5 and presented at the 61st annual meeting of the American Society of Hematology, Orlando, FL, 7-10 December 2019.
Data from the clinical trial will be made available after publication of the final analysis per request on an individual basis to comply with General Data Protection Regulation (GDPR) data privacy for the participating patients.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank all participating patients, local data managers for study coordination and patient data collection, local investigators, and, in particular, the study team at the HOVON Data Center at the Erasmus MC Cancer Institute and the 2 central laboratories at Amsterdam UMC and Rigshospitalet.
The work was supported by AbbVie and Janssen who also provided study drugs and had the option to comment on the manuscript.
The investigators are independent data owners with unrestricted publication rights.
Authorship
Contribution: A.P.K., M.-D.L., K.N., and C.U.N. designed the trial with A.P.K. and C.U.N. as international principal investigators; J.A.D. and I.S. were responsible for central MRD assays; J.D. and C.U.N. were responsible for the central biobanks; H.T.T.T., M.M., A.P.K., J.R., and C.U.N. were national principal investigators; S.K., L.E., G.J.V., R.M., C.H.M.M., C.B.P., H.F., A.J., E.C.D., and M.B. were local investigators; A.P.K., M.-D.L., K.N., J.D., and C.U.N. wrote the manuscript; and all authors have contributed to and approved the final manuscript.
Conflict-of-interest disclosure: A.P.K. received research funding and consultancy fees from AbbVie and Janssen and a speaker’s fee from AbbVie. J.D. received research funding from AbbVie. C.U.N. received research funding from the Novo Nordisk Foundation (grant NNF16OC0019302) and research support, consultancy fees, and/or travel grants from AbbVie, Gilead, Janssen, Roche, CSL Behring, Genmab, Sunesis, and Acerta/Astra Zeneca outside of this work. M.-D.L. received travel support and consultancy fees from AbbVie, Janssen, and Roche. S.K. received travel support from Cellgene. H.F. received research funding from Novartis, Alexion, Gilead, and AbbVie outside of this work. H.T.T.T. received consultancy fees from AbbVie, Pfizer, and Janssen; speaker’s fees from Novartis and Janssen; and research funding from CSL Behring. The remaining authors declare no competing financial interests.
Correspondence: Carsten U. Niemann, Department of Hematology, Rigshospitalet, Copenhagen University Hospital, Building 4041, Blegdamsvej 9, DK-2100 Copenhagen, Denmark; e-mail: carsten.utoft.niemann@regionh.dk.

