Key Points
FL-derived EVs impact BM stromal cell phenotype and favor their B-cell–supportive activity.
The GEP of BM FL B cells supports a specific network of interactions with EV-primed BM stromal cells.

Abstract
Follicular lymphoma (FL) originates in the lymph nodes (LNs) and infiltrates bone marrow (BM) early in the course of the disease. BM FL B cells are characterized by a lower cytological grade, decreased proliferation, and a specific phenotypic and subclonal profile. Mesenchymal stromal cells (MSCs) obtained from FL BM display a specific gene expression profile (GEP), including enrichment for a lymphoid stromal cell signature, and an increased capacity to sustain FL B-cell growth. However, the mechanisms triggering the formation of the medullar FL permissive stromal niche have not been identified. In the current work, we demonstrate that FL B cells produce extracellular vesicles (EVs) that can be internalized by BM-MSCs, making them more efficient to support FL B-cell survival and quiescence. Accordingly, EVs purified from FL BM plasma activate transforming growth factor β–dependent and independent pathways in BM-MSCs and modify their GEP, triggering an upregulation of factors classically associated with hematopoietic stem cell niche, including CXCL12 and angiopoietin-1. Moreover, we provide the first characterization of BM FL B-cell GEP, allowing the definition of the landscape of molecular interactions they could engage with EV-primed BM-MSCs. This work identifies FL-derived EVs as putative mediators of BM stroma polarization and supports further investigation of their clinical interest for targeting the crosstalk between BM-MSCs and malignant B cells.
Introduction
Follicular lymphoma (FL) is the most common indolent B-cell lymphoma, developing as a disseminated disease infiltrating lymph nodes (LNs) and bone marrow (BM).1 Within invaded LNs, malignant germinal center (GC)–derived B cells are found admixed with various nonneoplastic cells, including follicular helper CD4+ T cells (Tfh cells), lymphoid stromal cells, and macrophages, organized in follicular structures mirroring normal GC.2,3 Indeed, FL is considered a B-cell malignancy strongly dependent on a GC-like permissive microenvironment exhibiting FL-specific features. FL-infiltrating lymphoid stromal cells have recently emerged as key drivers of the FL-supportive niche. In agreement, the commitment of mesenchymal progenitors toward lymphoid-like stromal cell differentiation in vitro, through a combination of tumor necrosis factor-α (TNF) and lymphotoxin-α1β2 (LT), is associated with an increased capacity to sustain FL B-cell survival.4 Moreover, FL stromal cells were shown to overexpress CXCL12 in situ, contributing to migration, adhesion, and activation of FL B cells.5 Such specific alteration is associated with an extensive remodeling of the LN stromal cell phenotype.5-7
Besides nodal sites, BM infiltration is found in 40% to 70% of FL patients at diagnosis, preferentially with paratrabecular focal localizations, and is an important adverse factor in FL clinical risk-stratification scores.8-10 Interestingly, BM FL B cells are characterized by a lower cytological grade, decreased proliferation, and a modified phenotype with reduced CD10 expression compared with LN FL B cells.11 Moreover, somatic hypermutation analysis and targeted deep sequencing have demonstrated that different FL B-cell subclones could be detected within LN vs BM and suggested that FL originates in the LN and infiltrates BM early in the course of the disease, allowing further accumulation of BM-specific mutations.11-13 How tumor B cells are engaged in a bidirectional crosstalk with the BM microenvironment and what are the specific features of BM FL B cells remain largely unknown. Mesenchymal stromal cells (MSCs) obtained from FL BM display a specific gene expression profile (GEP), including an enrichment for a lymphoid stromal cell signature, and an increased capacity to sustain FL B-cell growth compared with BM-MSCs obtained from healthy donors (HDs).14 In addition, CXCL12 is specifically induced in BM-MSCs infiltrating FL BM aggregates.5 Finally, FL BM-MSCs also contribute to the organization of the FL niche by recruiting protumoral monocytes and neutrophils through CCL2 and interleukin-8 (IL-8), respectively.14,15 Both IL-8 and CCL2 could be increased in HD BM-MSCs by contact with FL B cells, suggesting that a dynamic reciprocal cooperation program is activated between FL B cells and FL-infiltrating stromal cells.14,15 Importantly, the early driver mechanism triggering the formation of this medullar FL permissive niche has not been identified.
Extracellular vesicles (EVs) are heterogeneous lipid bilayer–enclosed particles released from normal and malignant cells that are involved in cell-cell communication by transferring their bioactive cargos to recipient cells and play key roles in cancer biology.16 Recent studies have demonstrated that EVs released by cancer cells can be incorporated by BM-MSCs and contribute to their reprogramming into tumor-supportive cancer-associated fibroblasts (CAFs). This is particularly the case with lymphoid malignancies developing within the BM, such as multiple myeloma and chronic lymphocytic leukemia.17-21 However, no data are currently available regarding the role and properties of EVs in FL. In the current work, we report that FL B cells produce EVs that can be internalized by BM-MSCs, making them more efficient to support FL B-cell survival. Moreover, this supportive phenotype is different from the one obtained under TNF/LT stimulation, in agreement with the engagement of different signaling pathways in BM-MSCs, and includes a strong upregulation of genes previously involved in the hematopoietic stem cell (HSC) niche. Finally, BM-MSC priming by FL-derived EVs promotes FL B cells with a quiescent phenotype mimicking those of BM FL B cells. Altogether, these data support a role of EVs in FL dissemination and drug resistance.
Patients, materials, and methods
Patients and samples
The research protocol was conducted under French legal guidelines with informed consent and was approved by the local ethics committee. LN and BM aspirates were obtained from patients with FL at diagnosis, and BM was also collected from age-matched patients undergoing cardiac surgery. BM plasma was collected by centrifugation and frozen at −80°C until use. Ex vivo–expanded BM-MSCs and LN-derived lymphoid stromal cells (LN-LSCs) were obtained as previously described.4,14,22 The GEP of BM-MSCs and LN-LSCs was studied at the end of passage 1, and BM-MSCs were used at passages 2 to 3 for functional studies. For functional studies, HD B cells from tonsils and primary FL B cells from LNs were purified using a B-cell Isolation Kit II (Miltenyi Biotec). Purified LN FL B cells were also used to stimulate BM-MSCs for 3 days as previously described4 before sorting of CD45− CD105+ BM-MSCs for GEP. For malignant B-cell GEP, CD20hiCD44loCD38+IgD−CD138−CD34− viable FL B cells were sorted from LNs and invaded BM using a FACSAria (BD Biosciences). The GC-derived lymphoma B-cell lines RL, SUDHL4, and DOHH2 were obtained from the German Collection of Microorganisms and Cell Cultures and were maintained in RPMI 1640 supplemented with 7% fetal calf serum (Gibco) depleted from residual EVs by filtering at 200 nm followed by a 16-hour ultracentrifugation at 100 000g.
EV purification
All experiments related to EV purification and characterization were performed according to the Minimal Information for the Studies of Extracellular vesicles guideline of the International Society for Extracellular Vesicles23 and are detailed in supplemental Methods (available on the Blood Web site).
Study of BM-MSC priming by EVs
Analysis of the impact of EVs on BM-MSC RNA sequencing (RNA-seq) and function and study of the underlying signaling pathways are detailed in supplemental Methods.
GEP analysis of Affymetrix microarrays
RNA was extracted from 8 FL BM-MSCs, 8 FL LN-LSCs, 6 HD BM-MSCs primed or not by primary FL B cells, 5 BM FL B cells, and 10 LN FL B cells using Qiazol lysis with DNase treatment (Qiagen), and RNA purity and integrity were checked using the Bioanalyzer 2100 before analysis on GeneChip HG-U133 Plus 2.0 oligonucleotide microarrays (Affymetrix). In addition, we reanalyzed the GEP of our previous cohort of 10 FL BM-MSCs, including 6 from histologically invaded BM and 4 from histologically noninvaded BM, compared with 6 age-matched HD BM-MSCs.14
Analysis of differentially expressed genes, weighted gene correlation network analysis (WGCNA), biological pathway enrichment analysis, and analysis of the similarities between BM-MSC GEP and mouse single-cell RNA-seq data are described in supplemental Methods together with the validation of differentially expressed genes by quantitative polymerase chain reaction (qPCR).
Cytokine quantification
ANGPT1, TGF-β1 (R&D Systems), and CXCL12 (Merck Millipore) were quantified on BM plasma by Luminex technology.
Analysis of the interactome between FL B cells and stromal cells
Interactome analysis was performed with the NicheNet ligand activity algorithm24 in order to investigate the interface between EV-primed BM-MSCs and BM FL B cells and between TNF/LT-primed BM-MSCs and LN FL B cells. For details, see supplemental Methods.
Statistics
Statistical tests and graphic representation of the tunable resistive pulse sensing (TRPS), cytometry, qPCR, and Luminex data were performed with Prism software version 5 (GraphPad Software). Statistical significance was determined using analysis of variance, nonparametric Wilcoxon test for matched pairs, or Mann-Whitney nonparametric U test as appropriate. Data are presented as the mean ± standard deviation (SD) or median depending on the statistical test used.
Results
FL-derived EVs are internalized by BM-MSCs
Accumulating evidence suggests that EVs are an important mechanism underlying intercellular communication within malignant cells and their microenvironment. We thus purified EVs from conditioned media of FL B-cell lines and primary FL B cells using serial ultracentrifugation. EV size and concentration were studied and ranged from 60 to 300 nm with a mode of 90 to 110 nm and from 4 × 108 to 2.3 × 1010 particles/104 malignant B cells (Figure 1 A-C). We further validated that purified FL-derived vesicles expressed conventional EV markers, including the endosome-associated protein TSG101 and the tetraspanin CD81, in the absence of the contamination markers Golgin and Calnexin (supplemental Figure 1). Importantly, confocal microscopy and flow cytometry experiments revealed that BM-MSCs efficiently took up dyed EVs derived from FL B cells (supplemental Figure 2), whereas no transfer was observed on incubation at 4°C (Figures 1D). In addition, the use of endocytosis inhibitors, including EDTA, sucrose, and cytochalasin, and the specific targeting of the clathrin- and caveolin-dependent pathways through the dynamin-2 inhibitor Dynasore strongly reduced incorporation of FL-derived EVs in BM-MSCs (Figure 1D). We previously demonstrated that BM-MSCs could support FL B-cell survival in vitro.4,14 To evaluate how FL-derived EVs could modify the tumor-supportive capacity of BM-MSCs, we performed coculture experiments using purified primary FL B cells. As expected, HD BM-MSCs efficiently sustained the survival of neoplastic B cells. Moreover, preliminary treatment by FL-derived EVs significantly reinforced this feeder effect (P < .01; Figure 1E). Altogether, these data demonstrate that FL-derived EVs trigger BM-MSCs toward a FL-supportive phenotype.
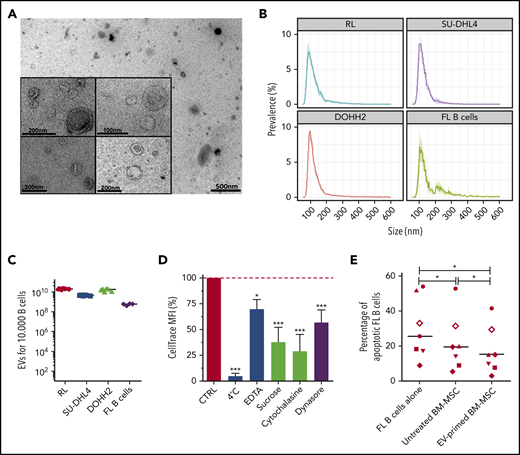
FL B cells produce EVs that are internalized by BM-MSCs and trigger their differentiation toward an FL-supportive stroma. (A) Transmission electron microscopy of EVs harvested by ultracentrifugation of conditioned medium from the RL B-cell line. Representative images of the data obtained with the different B-cell lines and primary FL B cells are shown. (B) Size analyze of EVs by TRPS. Data are represented as the mean ± SD from 6 (B-cell lines) or 4 (purified FL B cells) independent experiments. (C) Quantification of EVs by TRPS after 48 hours of culture of 104 B cells. (D) Flow cytometry analysis of 1.5 µM CellTrace Far Red–dyed EV uptake by BM-MSCs after 6 hours in the presence or not of specific endocytosis inhibitors. Data are represented as the mean ± SD of 4 independent experiments compared with the control condition (CTRL) arbitrarily assigned at 100%. Statistical significance was evaluated before normalization using the Dunnett post hoc test. ***P < .001; *P < .5. (E) Purified primary FL B cells (n = 8) were cocultured (ratio 4:1) or not with BM-MSCs pretreated or not with FL-derived EVs for 3 days. After 2 days, the percentage of active caspase-3+ CD19/CD20+ apoptotic malignant B cells was evaluated and compared with that of malignant B cells cultured alone using a nonparametric Wilcoxon test. Each symbol represents a different FL patient. *P < .05.
FL B cells produce EVs that are internalized by BM-MSCs and trigger their differentiation toward an FL-supportive stroma. (A) Transmission electron microscopy of EVs harvested by ultracentrifugation of conditioned medium from the RL B-cell line. Representative images of the data obtained with the different B-cell lines and primary FL B cells are shown. (B) Size analyze of EVs by TRPS. Data are represented as the mean ± SD from 6 (B-cell lines) or 4 (purified FL B cells) independent experiments. (C) Quantification of EVs by TRPS after 48 hours of culture of 104 B cells. (D) Flow cytometry analysis of 1.5 µM CellTrace Far Red–dyed EV uptake by BM-MSCs after 6 hours in the presence or not of specific endocytosis inhibitors. Data are represented as the mean ± SD of 4 independent experiments compared with the control condition (CTRL) arbitrarily assigned at 100%. Statistical significance was evaluated before normalization using the Dunnett post hoc test. ***P < .001; *P < .5. (E) Purified primary FL B cells (n = 8) were cocultured (ratio 4:1) or not with BM-MSCs pretreated or not with FL-derived EVs for 3 days. After 2 days, the percentage of active caspase-3+ CD19/CD20+ apoptotic malignant B cells was evaluated and compared with that of malignant B cells cultured alone using a nonparametric Wilcoxon test. Each symbol represents a different FL patient. *P < .05.
FL-derived EVs and TNF/LT differentially prime BM-MSCs toward an FL stroma-like phenotype
The priming of HD BM-MSCs by TNF/LT was shown to trigger an FL-supportive phenotype, and FL BM-MSCs have been described as ectopically committed toward lymphoid stroma differentiation.4,14 We thus decided to compare the GEP of 5 HD BM-MSCs stimulated by FL-derived EVs (obtained from 3 different FL B-cell lines) or TNF/LT, looking for molecular similarities and differences between these 2 types of induced tumor-promoting stroma. RNA-seq analysis revealed a lack of significant overlap between the signatures induced by EVs vs TNF/LT (P = 1, hypergeometric test; Figure 2A), delineating a specific EV signature (1218 upregulated genes) and a specific TNF/LT signature (1312 upregulated genes) (supplemental Table S2). In order to evaluate the biological relevance of these 2 signatures, we then tested whether they were enriched in FL-infiltrating stromal cells obtained from BM (FL BM-MSCs) or LN (FL LN-LSCs). For that purpose, we examined the GEP of FL BM-MSC (n = 8) and FL LN-LSC (n = 8) using Affymetrix microarrays. Unsupervised principal-component analysis (PCA) performed on all expressed genes adequately segregated the 2 FL stromal cell subsets (Figure 2B) and we identified 1050 genes specifically upregulated in FL BM-MSCs and 874 genes specifically upregulated in FL LN-LSC (supplemental Table S3). Interestingly, gene set enrichment analysis (GSEA) revealed that whereas the TNF/LT signature was enriched in FL LN-LSCs, in agreement with their localization within heavily infiltrated lymphoid tissues, the FL BM-MSC transcriptomic profile was strongly enriched for the EV signature, raising the hypothesis that FL-derived EVs could contribute to the activation of stromal cells outside the primary LN niches (Figure 2C). To extend these data, we took the opportunity to reanalyze our previous GEP cohort of 10 FL BM-MSCs, including 6 from histologically invaded BM and 4 from histologically noninvaded BM, compared with 6 age-matched HD BM-MSCs.14 First, a PCA performed on the 2389 genes differentially expressed between FL BM-MSCs and HD BM-MSCs (false discovery rate <1%) suggested that BM-MSCs obtained from invaded BM could be distinguished from those obtained from noninvaded BM (supplemental Figure 3A). Moreover, the TNF/LT signature, unlike the EV signature, was enriched in BM-MSCs in close contact with FL B cells within invaded BM, confirming that the TNF/LT signature, unlike the EV signature, was related to direct contact of BM-MSCs with malignant B cells (supplemental Figure 3B). To further understand the direct impact of malignant B cells on the BM-MSC phenotype, we performed 3-day cocultures (n = 6) of purified primary FL B cells with HD BM-MSCs and analyzed the GEP of cell-sorted primed BM-MSCs (supplemental Table S4). Importantly, GSEA highlighted that this 1716-gene signature was significantly enriched in FL BM-MSCs compared with HD BM-MSCs, thus validating the relevance of our in vitro approach (supplemental Figure 3C). To decipher the relationship between the BM-MSC phenotype induced by FL-derived EV vs FL B cells, we then delineated the specific FL B-cell primed signature (1551 upregulated genes; supplemental Figure 3D). Interestingly, whereas this signature was not enriched in FL BM-MSCs vs FL LN-LSCs, it was strongly enriched in BM-MSCs obtained from invaded vs noninvaded BM (supplemental Figure 3E-F), further suggesting a role of EVs in the activation of BM-MSCs before BM seeding by malignant B cells.
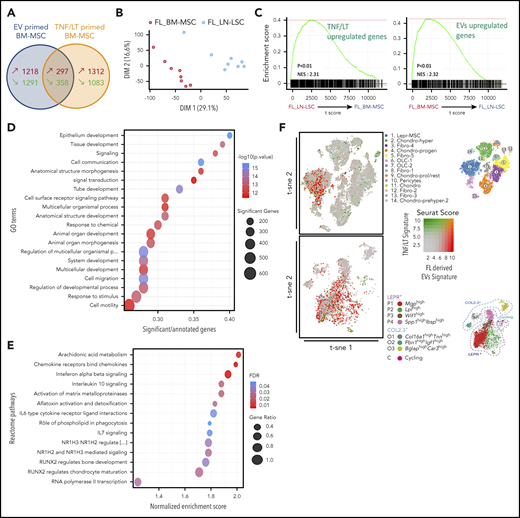
Comparison of the GEP of BM-MSCs primed by FL-derived EVs or TNF/LT. (A) Venn diagram representing the number of genes upregulated and downregulated in HD BM-MSCs (n = 5) by FL-derived EVs from 3 different FL B-cell lines vs TNF/LT, as evaluated by RNA-seq. (B) Unsupervised PCA of Affymetrix GEP of FL-derived BM-MSCs (FL_BM-MSC) (n = 8) and FL-derived LN-LSCs (FL_LN-LSC) (n = 8). (C) GSEA enrichment scores for the TNF/LT signature (1312 upregulated genes) and EV signature (1218 upregulated genes) between FL_BM-MSCs and FL_LN-LSCs. (D-E) Study of the EV signature using GO term enrichment analysis (D) and GSEA on REACTOME pathways (E). (F) Comparison of the EV-primed BM-MSCs and the TNF/LT-primed BM-MSCs with the murine BM mesenchymal cells recently described by single-cell RNA-seq.25,26 The assignment of the mouse BM stroma cell clusters restricted to the mesenchymal compartments as described in these 2 studies is indicated on the right. FDR, false discovery rate; t-sne, t-distributed stochastic neighbor embedding.
Comparison of the GEP of BM-MSCs primed by FL-derived EVs or TNF/LT. (A) Venn diagram representing the number of genes upregulated and downregulated in HD BM-MSCs (n = 5) by FL-derived EVs from 3 different FL B-cell lines vs TNF/LT, as evaluated by RNA-seq. (B) Unsupervised PCA of Affymetrix GEP of FL-derived BM-MSCs (FL_BM-MSC) (n = 8) and FL-derived LN-LSCs (FL_LN-LSC) (n = 8). (C) GSEA enrichment scores for the TNF/LT signature (1312 upregulated genes) and EV signature (1218 upregulated genes) between FL_BM-MSCs and FL_LN-LSCs. (D-E) Study of the EV signature using GO term enrichment analysis (D) and GSEA on REACTOME pathways (E). (F) Comparison of the EV-primed BM-MSCs and the TNF/LT-primed BM-MSCs with the murine BM mesenchymal cells recently described by single-cell RNA-seq.25,26 The assignment of the mouse BM stroma cell clusters restricted to the mesenchymal compartments as described in these 2 studies is indicated on the right. FDR, false discovery rate; t-sne, t-distributed stochastic neighbor embedding.
To explore the EV signature more deeply, we next submitted it to pathway enrichment analysis using Gene Ontology (GO), REACTOME, and MSigDB databases (Figure 2D-E; supplemental Table S5). Whereas the TNF/LT signature was associated to inflammation- and NF-κB–related pathways (supplemental Figure 4 and supplemental Table S5), the EV signature was enriched for cell signaling and cell communication biological processes, cytokine/chemokine signaling (including IL-6, IL-7, and transforming growth factor β [TGF-β]), and pathways related to bone and cartilage development. This result prompted us to compare our data to the recently published mouse data sets describing the heterogeneity of steady state BM-MSC.25,26 Interestingly, BM-MSCs stimulated by FL-derived EVs harbored a GEP close to that of leptin receptor–positive (Lepr)+ MSCs and MSC-derived early osteolineage cells (OLCs), both reported to be key cell subsets of HSC niche (Figure 2F). Conversely, the signature of TNF/LT-primed BM-MSCs did not match a specific mouse BM stromal cell subset. Altogether, these data demonstrate that FL-derived EVs trigger a particular phenotype in HD BM-MSCs, sharing features with FL-infiltrating stromal cells and HSC stromal niche.
BM-MSCs stimulated by FL-derived EVs upregulate genes of the HSC and FL BM niche
Detailed analysis of the EV-primed BM-MSC signature highlighted all the master genes involved in HSC maintenance and regulation. In particular, CXCL12, angiopoietin-1 (ANGPT1), angiopoietin-2 (ANGPT2), KITLG/SCF, and EPHB4 were upregulated by FL-derived EVs, similarly to IL7, a key factor of early commitment toward B-cell lineage that was recently shown to be coexpressed in perisinusoidal cells with CXCL12 and KITLG/SCF27 (Figure 3A). Apart from these functional markers, FL-derived EVs did not trigger the classical markers of MSC compartment, such as LEPR, NES, NG2, or CD200,26 but upregulated markers of osteolineage differentiation, including RUNX2, the master regulator transcription factor controlling the commitment of MSCs to OLCs,28 and SP7, its downstream target. The lack of induction of mature OLC markers, including BGLAP, ALPL, or SPP1, argued for an osteo-primed MSC stage rather than a full osteoblastic differentiation. In agreement, repeated stimulation of HD BM-MSCs with FL-derived EVs triggered partial osteoblastic differentiation compared with that obtained with classical osteoblastic differentiation medium (supplemental Figure 5). Moreover, the adipogenic marker PPARG was also upregulated in response to stimulation by FL-derived EVs, generating a phenotype close to that of the Lepr+ cells of the HSC niche that coexpress a unique combination of adipogenic and osteogenic transcription factors.29 Finally, both FOXC1 and EBF1, the 2 transcription factors essential for the formation and maintenance of HSC niche and the inhibition of full adipocyte and osteoblast differentiation,30,31 were upregulated by FL-derived EVs in BM-MSCs. Collectively, EV-primed BM-MSCs expressed the specific panel of transcription factors, cytokines, and chemokines previously reported in HSC stromal cell niches. Of note, RUNX2 and EPHB4 were both also upregulated by priming of BM-MSCs with primary FL B cells (supplemental Figure 3D). Conversely, the TNF/LT-induced signatures associated with a decreased expression of CXCL12, ANGPT2, and RUNX2, together with an upregulation of inflammatory cytokines, including IL1A, IL1B, IL6, IL15, inflammatory chemokines, such as CCL2, CXCL8, CXCL10, and CX3CL1, and the adhesion molecules ICAM1, VCAM1, and ITGAV (Figure 3A). Of note, TGF-β signaling has been identified as regulating HSC quiescence in the BM niche, and both TGFB1 and TGFB3 were significantly upregulated in EV-primed vs TNF/LT-primed BM-MSCs. All these data were confirmed by qPCR in BM-MSCs treated by TNF/LT (n = 9) vs FL-derived EVs (n = 27) (Figure 3B).
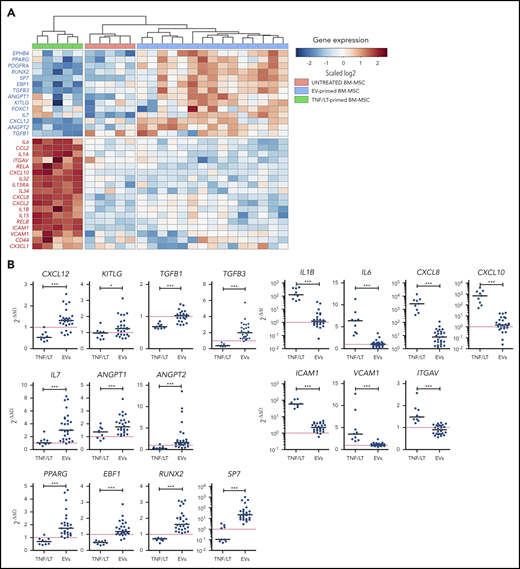
HSC niche signature is enriched in EV-primed BM-MSCs. (A) Hierarchical clustering of untreated, TNF/LT-primed, and EV-primed BM-MSCs on selected differentially expressed genes as evaluated by RNA-seq. The relative level of gene expression is depicted according to the shown color scale. (B) qPCR validation of the RNA-seq data for BM-MSCs treated by FL-derived EVs (n = 27) and TNF/LT (n = 9) highlighting genes upregulated by EVs (left) or TNF/LT (right). Results are expressed as the fold change compared with untreated BM-MSCs assigned at 1 (red dotted line). *P < .05; ***P < .001.
HSC niche signature is enriched in EV-primed BM-MSCs. (A) Hierarchical clustering of untreated, TNF/LT-primed, and EV-primed BM-MSCs on selected differentially expressed genes as evaluated by RNA-seq. The relative level of gene expression is depicted according to the shown color scale. (B) qPCR validation of the RNA-seq data for BM-MSCs treated by FL-derived EVs (n = 27) and TNF/LT (n = 9) highlighting genes upregulated by EVs (left) or TNF/LT (right). Results are expressed as the fold change compared with untreated BM-MSCs assigned at 1 (red dotted line). *P < .05; ***P < .001.
To evaluate the relevance of these observations for the FL BM niche, we stimulated BM-MSCs with EVs isolated from BM plasma of FL patients (n = 13) vs age-matched HDs (n = 6) and characterized by transmission electron microscopy and TRPS (Figure 4A-B). Interestingly, the hematopoietic cytokines CXCL12, ANGPT1, TGFB3, and IL7, as well as the master regulators of HSC niche formation EBF1 and FOXC1, were significantly induced by EVs obtained from FL BM compared with HD BM, suggesting that the polarization process observed in vitro could take place within FL BM niches (Figure 4C). In addition, whereas the level of IL-7 and TGF-β3 remained below the quantification level in all BM plasma tested, we could extend our previous demonstration of CXCL12 upregulation within FL BM niche5 and identified a significant increase of angiopoietin-1 and TGF-β1 levels in FL BM plasma (Figure 4D).
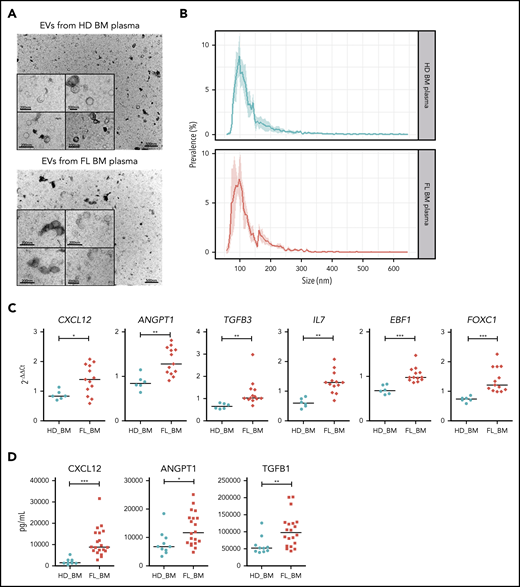
Characterization of the phenotype of BM-MSC treated by EVs purified from FL BM plasma. (A) Representative image of transmission electron microscopy of EVs harvested by ultracentrifugation of HD BM plasma (upper) and FL BM plasma (bottom). (B) Size analyze by TRPS of EVs harvested from HD BM plasma (upper, n = 4) or FL BM plasma (bottom, n = 4). Data are represented as the mean ± SD. (C) qPCR of BM-MSCs treated by 15 µL EVs freshly enriched from BM plasma of HD (n = 6) and FL patients (n = 13). Results expressed as fold change of untreated BM-MSCs. *P < .05; ** P < .01. (D) Luminex assay on BM plasma from HD BM (n = 10) and FL BM (n = 20). P < .05, **P < .01, ***P < .001.
Characterization of the phenotype of BM-MSC treated by EVs purified from FL BM plasma. (A) Representative image of transmission electron microscopy of EVs harvested by ultracentrifugation of HD BM plasma (upper) and FL BM plasma (bottom). (B) Size analyze by TRPS of EVs harvested from HD BM plasma (upper, n = 4) or FL BM plasma (bottom, n = 4). Data are represented as the mean ± SD. (C) qPCR of BM-MSCs treated by 15 µL EVs freshly enriched from BM plasma of HD (n = 6) and FL patients (n = 13). Results expressed as fold change of untreated BM-MSCs. *P < .05; ** P < .01. (D) Luminex assay on BM plasma from HD BM (n = 10) and FL BM (n = 20). P < .05, **P < .01, ***P < .001.
FL-derived EVs activated various signaling pathways in BM-MSC
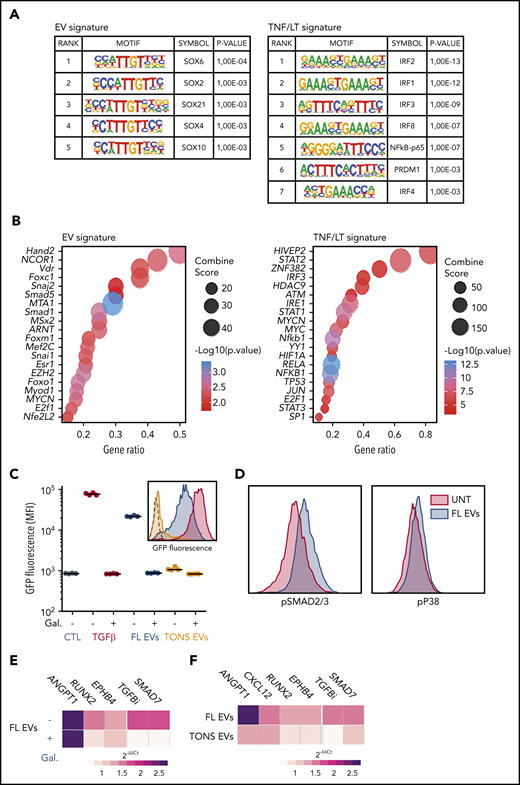
We next tried to identify the signaling pathways activated by FL-derived EVs in BM-MSC. For that purpose, we first evaluated the enrichment for transcription factor motifs in the promoters of genes upregulated in EV vs TNF/LT signatures and the enrichment for transcription factor targets in these 2 signatures (Figure 5A-B). The TNF/LT signature was in particular associated with NF-κB and interferon regulatory factor signaling pathways, whereas the EV signature revealed activity of SOXs, SMADs, MTA1, or EZH2 transcription factors, all previously reported in the TGF-β signaling pathway.32 Interestingly, besides numerous proteins related to B-cell activation, including BTK, CD81, CD79A, MME, or major histocompatibility complex class II, quantitative proteomics of EVs by liquid chromatography-mass spectrometry revealed the presence of higher levels of TGF-β1 in FL B-cell–derived EVs compared with tonsil B-cell–derived EVs (supplemental Table S6). In agreement, FL-derived EVs did not trigger the nuclear translocation of NF-κB p65 subunit in HD BM-MSCs (supplemental Figure 6A) but induced activation of SMAD2/3-SMAD4 in a TGF-β sensor cell line (Figure 5C). Of note, SMAD activation by FL-derived EVs was specifically reversed by the TGFBR1 inhibitor galunisertib and was not induced by tonsil B-cell–derived EVs, in agreement with the upregulation of TGFB1 recently reported in primary purified FL B cells compared with normal GC B cells.33 Finally, FL-derived EVs activated SMAD2/3 and p38 pathways related to canonical and noncanonical TGF-β signaling in BM-MSCs (Figure 5D). Importantly, galunisertib inhibited only partly the activity of FL-derived EVs as highlighted by the reduction of RUNX2 and EPHB4, but not ANGPT1, expression (Figure 5E). This could be related to the fact that FL-derived EVs also triggered non-TGF-β–related signaling pathways, including activation of STAT6, unlike STAT1/3/5, phosphorylation (supplemental Figure 6B). STAT6 was also identified within the proteins enriched in FL-derived EVs compared with tonsil B-cell–derived EVs (supplemental Table S6), reinforcing the hypothesis that FL-derived EVs trigger STAT6 activation in BM-MSCs. Altogether, these data suggest that FL-derived EVs activated TGF-β–dependent and TGF-β–independent activation pathways in BM-MSCs that are essentially not triggered by tonsil-derived EVs, as confirmed at the transcriptomic level (Figure 5F).
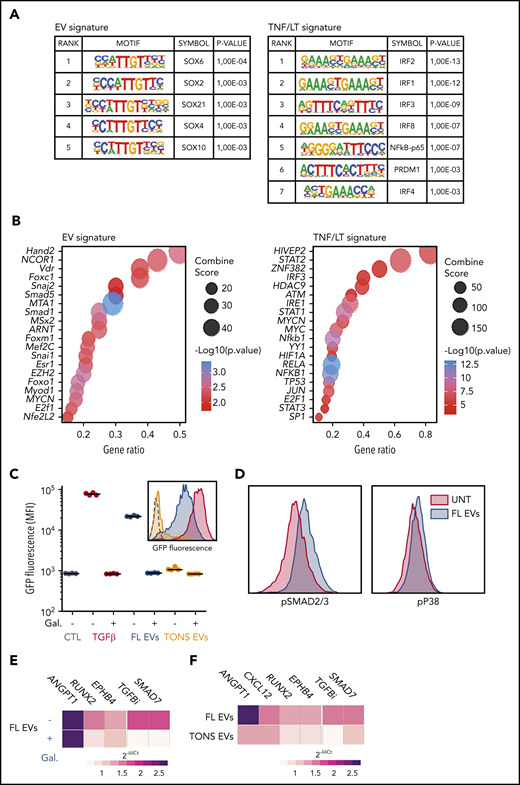
EV-induced signalization in BM-MSCs. (A) Analysis of transcription factor enrichment in the promoters of EV (left) or TNF/LT (right) upregulated genes performed with HOMER. Represented are significant motifs. (B) Enrichment for transcription factor targets in EV (left) or TNF/LT (right) upregulated genes as determined with TRRUST database with EnrichR Web site. The Top20 enriched transcription factors were represented as dot plots. (C) A TGF-β sensor cell line was stimulated overnight by TGF-β1, EVs from the RL cell line (FL EVs), or pooled EVs from tonsil B cells (TONS EVs) in the presence or not of the TGFBR1 inhibitor galunisertib (Gal). GFP expression was determined by flow cytometry and expressed as the mean fluorescence intensity (MFI). Shown are 4 independent experiments with 1 example of a representative data in the histogram. (D) Evaluation of SMAD2/3 and p38 phosphorylation by flow cytometry after a 6-hour stimulation by RL EVs in comparison with untreated (UNT) cells. Shown is one representative experiment out of 3. (E) Analysis by qPCR of BM-MSCs (n = 6) treated by RL EVs ± TGFBR1 inhibition with galunisertib. Results are expressed as median fold change of untreated BM-MSCs. (F) Analysis by qPCR of BM-MSCs (n = 6) treated with EVs from purified tonsil B cells. Results are expressed as median fold change of untreated BM-MSCs.
EV-induced signalization in BM-MSCs. (A) Analysis of transcription factor enrichment in the promoters of EV (left) or TNF/LT (right) upregulated genes performed with HOMER. Represented are significant motifs. (B) Enrichment for transcription factor targets in EV (left) or TNF/LT (right) upregulated genes as determined with TRRUST database with EnrichR Web site. The Top20 enriched transcription factors were represented as dot plots. (C) A TGF-β sensor cell line was stimulated overnight by TGF-β1, EVs from the RL cell line (FL EVs), or pooled EVs from tonsil B cells (TONS EVs) in the presence or not of the TGFBR1 inhibitor galunisertib (Gal). GFP expression was determined by flow cytometry and expressed as the mean fluorescence intensity (MFI). Shown are 4 independent experiments with 1 example of a representative data in the histogram. (D) Evaluation of SMAD2/3 and p38 phosphorylation by flow cytometry after a 6-hour stimulation by RL EVs in comparison with untreated (UNT) cells. Shown is one representative experiment out of 3. (E) Analysis by qPCR of BM-MSCs (n = 6) treated by RL EVs ± TGFBR1 inhibition with galunisertib. Results are expressed as median fold change of untreated BM-MSCs. (F) Analysis by qPCR of BM-MSCs (n = 6) treated with EVs from purified tonsil B cells. Results are expressed as median fold change of untreated BM-MSCs.
BM FL B cells specifically interact with BM-MSCs primed by FL-derived EVs
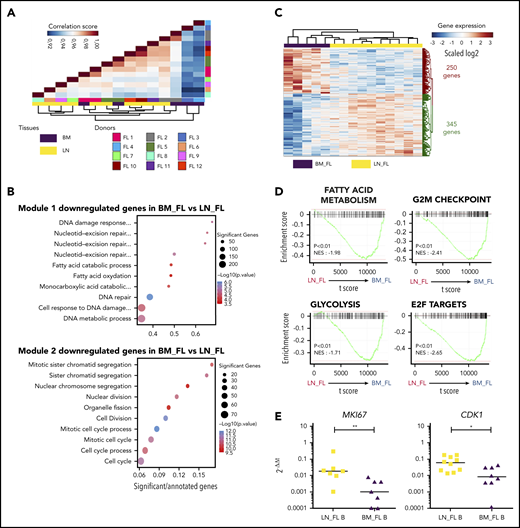
Having demonstrated that LN and BM stromal cells from FL patients displayed different GEPs enriched for TNF/LT vs EVs signatures, respectively, we wondered whether FL B cells purified from LNs vs BM also exhibited specific molecular features. For that purpose, we analyzed the transcriptome of sorted CD20hiCD44loCD38+IgD−CD138−CD34− B cells obtained from invaded LNs (n = 10) and BM (n = 5). The first parameter impacting FL B-cell GEP was interindividual variability, as highlighted by the close relationship between autologous LN and BM B cells in unsupervised Pearson correlation (Figure 6A). We then applied an unsupervised WGCNA to identify transcriptional modules of coexpressed genes and evaluate whether they were related to FL B-cell origin. The coexpression network constructed by using the WGCNA algorithm allowed us to identify 17 distinct gene modules (supplemental Figure 7A). We then determined whether the module eigengenes were significantly different in BM vs LN FL B cells (supplemental Figure 7B). Two modules (Blue_module 1 and Brown_module 2) were significantly associated with the FL B-cell origin, and the top 10 GO terms associated with both of them highlighted a downregulation of genes involved in cell cycle and fatty acid metabolism in BM FL B cells, indicating that interacting with a different microenvironment impacted FL B-cell profile (Figure 6B). We next identified the 595 differentially expressed genes between LN and BM FL B cells and revealed by GSEA that BM FL B-cell GEPs reflected a reduced proliferation and active metabolism (Figure 6C-D). In agreement, BM FL B cells overexpressed the antiproliferative genes BTG1 (previously involved in T-cell and HSC quiescence34,35 ), CKAP4, and LZTS3,36 but also Dysadherin/FXYD5, a molecule involved in drug resistance and cell motility in cancer stem cells37 (supplemental Table S7). Finally, we confirmed by qPCR the lower expression of MKI67 and CDK1 in BM FL B cells vs LN FL B cells (Figure 6E).
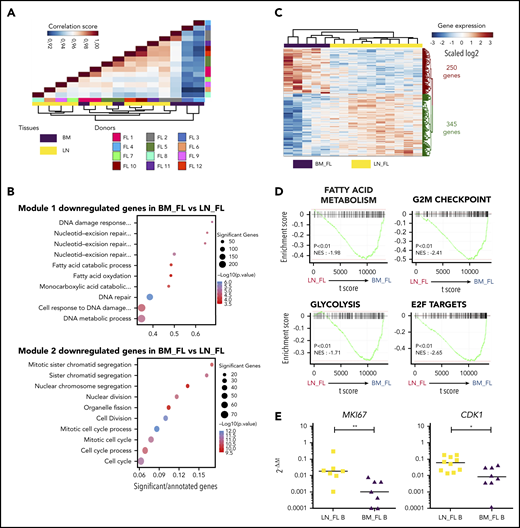
Characterization of BM FL B-cell phenotype. (A) Unsupervised Pearson correlation clustering on all expressed genes in purified FL B cells from LNs (n = 10) and BM (n = 5), including 3 paired BM/LN samples (FL1, FL2, and FL5). (B) List of the top 10 GO terms associated with the 2 modules identified as correlated with the tissue origin of FL B cells by WGCNA. (C) Heatmap of the differentially expressed genes between BM FL B cells and LN FL B cells (P < .01). (D) GSEA of the MSigDB hallmark pathways comparing BM FL B cells and LN FL B cells. (E) qPCR validation of the downregulation of MKI67 and CKD1 in BM FL B cells (n = 7) compared with LN FL B cells (n = 10). Results are represented in arbitrary units obtained by assigning the value of 1 to a pool of PBMCs. *P < .05; **P < .01
Characterization of BM FL B-cell phenotype. (A) Unsupervised Pearson correlation clustering on all expressed genes in purified FL B cells from LNs (n = 10) and BM (n = 5), including 3 paired BM/LN samples (FL1, FL2, and FL5). (B) List of the top 10 GO terms associated with the 2 modules identified as correlated with the tissue origin of FL B cells by WGCNA. (C) Heatmap of the differentially expressed genes between BM FL B cells and LN FL B cells (P < .01). (D) GSEA of the MSigDB hallmark pathways comparing BM FL B cells and LN FL B cells. (E) qPCR validation of the downregulation of MKI67 and CKD1 in BM FL B cells (n = 7) compared with LN FL B cells (n = 10). Results are represented in arbitrary units obtained by assigning the value of 1 to a pool of PBMCs. *P < .05; **P < .01
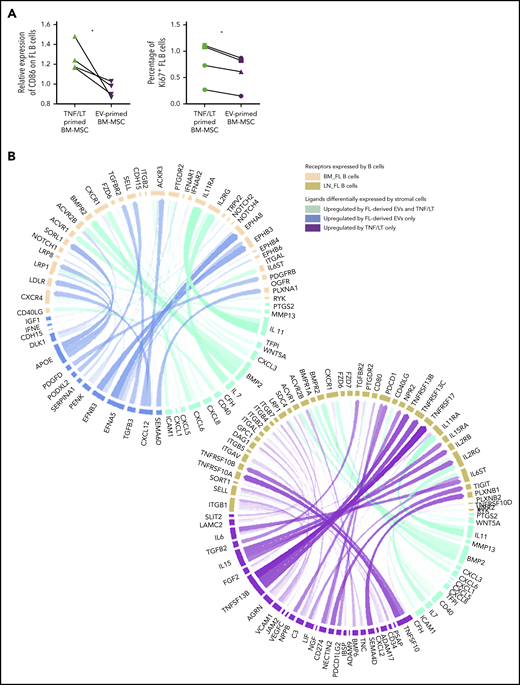
Given the specific similarities between EV-primed BM-MSCs and BM FL stroma, we then evaluated the impact of EV-primed BM-MSCs on primary FL B cells purified from invaded LNs compared with TNF/LT-primed BM-MSCs as a reference of lymphoid-committed stromal cells (Figure 7A). Interestingly, FL B cells cocultured with EV-primed BM-MSCs exhibited a decreased expression of CD86 and a lower rate of proliferation, without a difference in cell survival compared with coculture with TNF/LT-derived BM-MSCs (data not shown). Finally, based on our 2 FL-supportive stromal cell signatures and the list of genes upregulated in BM and LN FL B cells compared with normal centrocytes38 (supplemental Table S8), we next applied the new computational method NicheNet24 to delineate the specific interactions underlying the crosstalk between FL B cells and stromal cells, defining stromal cells as sender cells and B cells as receiver cells (Figure 7B). Using this strategy, we highlighted, as expected, CXCL12 and TGFB3 at the specific interface between BM FL B cells and EV-primed BM-MSC, and we also identified several members of the Eph/ephrin receptor family known to mediate cell-cell signals in a variety of physiological contexts. Conversely, the crosstalk between LN FL B cells and TNF/LT-primed BM-MSCs specifically involved IL-6, IL-15, and TNFSF13B/BAFF pathways.
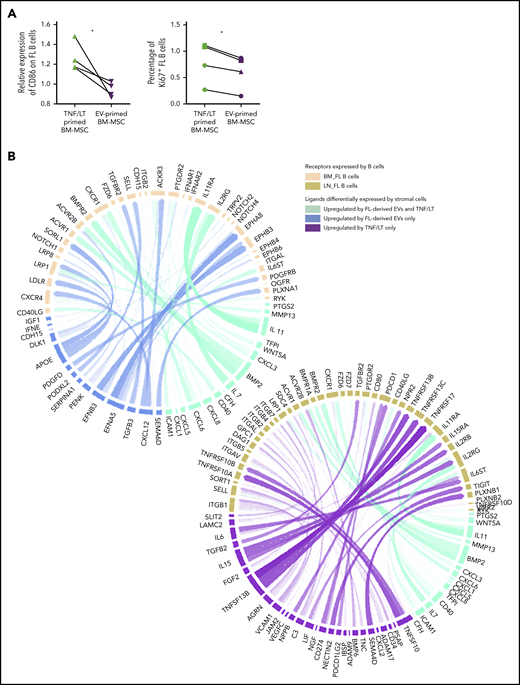
Characterization of the interaction of BM FL B cells with stromal cells. (A) Purified FL B cells (n = 4) were cocultured with BM-MSCs pretreated with TNF/LT or FL-derived EVs for 3 days. On day 3, CD19/CD20+ viable B cells were analyzed by flow cytometry for CD86 membrane expression (left; expressed as a ratio of mean fluorescence intensity compared with B cells cocultured on untreated BM-MSCs) and Ki67 nuclear expression (right; expressed as a percentage of stained B cells). (B) Circos plots showing predicted interactions between BM-MSCs defined as sender cells and FL B cells defined as receiver cells based on the genes enriched in the EV signature and/or TNF/LT signature compared with untreated BM-MSCs and genes enriched in BM FL B cells (left) or LN FL B cells (right) compared with normal centrocytes.
Characterization of the interaction of BM FL B cells with stromal cells. (A) Purified FL B cells (n = 4) were cocultured with BM-MSCs pretreated with TNF/LT or FL-derived EVs for 3 days. On day 3, CD19/CD20+ viable B cells were analyzed by flow cytometry for CD86 membrane expression (left; expressed as a ratio of mean fluorescence intensity compared with B cells cocultured on untreated BM-MSCs) and Ki67 nuclear expression (right; expressed as a percentage of stained B cells). (B) Circos plots showing predicted interactions between BM-MSCs defined as sender cells and FL B cells defined as receiver cells based on the genes enriched in the EV signature and/or TNF/LT signature compared with untreated BM-MSCs and genes enriched in BM FL B cells (left) or LN FL B cells (right) compared with normal centrocytes.
Discussion
FL is characterized by a frequent BM infiltration by malignant B cells displaying BM-specific phenotypic and genetic features associated with an alteration of the local immune and stromal microenvironment, supporting a key role for bidirectional interactions between FL cells and their surrounding medullar niche.39-41 The specific gene signature of FL BM stromal cells was initially identified as independent of the infiltration extend,14 suggesting additional mechanisms of stroma polarization besides direct cell-cell contact. Several reports have demonstrated that EVs released by malignant cells from primary solid tumors can modify distal sites, including the BM, to promote metastasis.42,43 In the current study, we thus explored the relationship between FL BM stroma and FL BM B-cell features and how FL-derived EVs could contribute to the commitment of BM-MSCs toward a lymphoma-supportive stroma.
We first demonstrated that FL-derived EVs strongly modified the GEP of BM-MSCs, triggering an upregulation of HSC niche factors, including CXCL12. Such induction of CXCL12 in the BM stroma by tumor EVs has been recently described in prostate cancers and was shown to be dependent on the upregulation of HIF-1α,43 which is also induced in BM-MSCs by FL-derived EVs (supplemental Table S2). CXCL12 supports FL B-cell migration, adhesion, and survival5 and could thus be involved in the homing and growth of BM FL B cells. Of note FL-derived EVs also triggered angiopoetin-1 in BM-MSCs, and we confirmed its increased amount in FL BM plasma. In the context of myeloid neoplasms, tumor EVs repress angiopoietin-1 expression,44 demonstrating that supportive stroma phenotypes are tumor specific. Collectively, the aberrant expression of hematopoietic niche factors in FL BM could contribute to the deregulated hematopoiesis described in patients with mature B-cell malignancies even in the absence of detectable BM involvement.45 Importantly, EVs could also have an impact on the immune compartments of the tumor microenvironment. In particular, tumor-derived EVs have been involved in the polarization of tumor-associated macrophages in Waldenstrom macroglubulinemia46 and diffuse large B-cell lymphoma, the most frequent aggressive B-cell lymphoma, in which EV-primed macrophages were shown to sustain malignant B-cell proliferation and migration.47 Given the key role of tumor-associated macrophages in FL pathogenesis, it would be interesting to evaluate how FL-derived EVs contribute to their reprogramming. Finally, we cannot exclude that tumor EVs directly favor malignant B-cell growth through the delivery of FL B-cell growth factors, as suggested for the activity of IL-31.48
EVs from Hodgkin lymphoma promote a proinflammatory and proangiogenic NF-κB–dependent phenotype in fibroblasts.49 Conversely, stroma reprogramming by FL-derived EVs seems to be NF-kB independent and is not associated with activation of inflammatory pathways. However, malignant FL B cells could also directly affect the phenotype and function of infiltrating stromal cells, and the release of TNF/LT by tumor B cells is involved in the upregulation of CCL2 and IL-8 in BM and LN FL stroma.14,15 The net effect of EVs vs direct contact with tumor cells on the phenotype of FL stromal cells probably depends on both the tumor site (ie, primary LN vs distant BM) and the kinetics of lymphoma evolution, with an increase in BM involvement in more advanced stages of the disease. Of note, when BM-MSCs were treated by a combination of TNF/LT and FL-derived EVs, CXCL12 was downregulated, indicating a dominant effect of contact with B cells (data not shown), thus explaining the discrepancy between BM and LN stroma phenotypes. Besides malignant B cells, the FL tumor microenvironment could contribute to FL stroma polarization. This is particularly the case for FL-infiltrating Tfh cells that overexpress IL-4 and promote CXCL12 expression in FL LN stromal cells.5 Interestingly, stimulation of BM-MSCs by FL-derived EVs induced an upregulation of IL4R, STAT6, and CD40 (supplemental Figure S8), all of which are involved in the response to IL-4– and CD40L–expressing T cells, and FL-derived EVs were enriched for STAT6 protein. Importantly, very few T cells with a full Tfh phenotype could be detected in FL BM, and CD4/CD8 ratio was found increased only in heavily involved BM, whereas the level of CXCL12 is already increased in noninvolved BM plasma. These data suggested that EVs could contribute to CXCL12 upregulation in the absence of direct contact with malignant B cells but could then synergize with IL-4 produced by infiltrating T cells admixed with FL B cells.5
EVs are known to trigger complex signaling networks in CAF related to the delivery of numerous proteins, RNA, and lipids. Interestingly, we highlighted a TGF-β–dependent activation of BM-MSCs by FL-derived EVs. Expression of TGF-β by LN FL B cells has been previously described and was involved in the alteration of intratumoral T-cell function.50,51 More recently, an integrative analysis of the FL cell niche revealed enrichment for gene networks related to TGF-β signaling.33 How TGF-β and STAT6 pathways synergize for the acquisition of an FL CAF phenotype remains to be explored. These data, together with our current demonstration that FL-derived EVs contribute to the TGF-β–dependent activation of distant BM-MSCs, support TGF-β as a putative target deserving further investigation.
FL BM-MSCs support malignant B cells with a lower proliferation rate than LN FL B cells.10 In the current study, we provide the first characterization of BM FL B-cell GEPs and confirmed their quiescent phenotype associated with upregulation of the antiproliferative gene BTG1. Interestingly, a BTG1hi phenotype was recently associated with a good prognosis in aggressive diffuse large B-cell lymphoma,52 in association with a decreased cell cycle, confirming a role for BTG1 in the regulation of cell proliferation in GC-derived B-cell lymphomas. The analysis of the interactome between BM FL B cells and EVs-primed BM-MSCs highlights not only CXCL12 but also the Eph/ephrin pathway. Silencing EphA7 receptor, a decoy receptor blocking Eph signaling, has been shown to drive lymphoma development in a murine FL model,53 suggesting that ephrin-dependent activation could have a protumoral activity in FL.
Our study suggests that FL-derived EVs should be considered as putative mediators of BM stroma polarization. Moreover, EVs released by lymphoma B cells were already demonstrated to bind therapeutic anti-CD20 antibodies, protecting malignant cells from antibody attack.54 In this context, the blockade of EV release was proposed as clinically relevant to restore sensitivity to immunotherapy. Whether FL pathogenesis might also be influenced by EVs released by the tumor microenvironment, as previously reported for other hematopoietic malignancies,55 remains to be elucidated. Besides the mechanism of BM stroma reprogramming, BM forms a survival niche supporting resistance to treatment and inducing LN relapses12 in FL, supporting the clinical interest of targeting the crosstalk between BM-MSCs and malignant B cells, either through the inhibition of stroma-derived B-cell growth factors or through the mobilization of FL B cells outside their supportive BM niche.
Data are available under data set 1 (GSE152386), data set 2 (GSE152386 and GSE152068), data set 3 (GSE35331), and data set 4 (GSE152215, GSE85233, and GSE136249) in the GEO database.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
There is a Blood Commentary on this article in this issue.
Acknowledgments
The authors thank the personnel of Carte d’Identité des Tumeurs platforms for qualification (Saint-Louis Hospital, Paris, France) and Affymetrix expression arrays (Institut de Génétique et de Biologie Moléculaire et Cellulaire, Strasbourg, France). The authors are indebted to the Centre de Ressources Biologiques Santé (BB-0033-00056; http://www.crbsante-rennes.com) of Rennes Hospital for its support in the processing of biological samples.
This work was supported by research grants from Fondation ARC (PGA1 RF20170205386), the Institut National du Cancer (INCA AAP PNP19-009), the Infrastructure Program EcellFRANCE (ANR-11-INSB-005), and was part of the Carte d’Identité des Tumeurs Program developed by the Ligue contre le Cancer. S.L. is supported by the Rennes 1 Foundation “Cancer & Innovation” Chair, and by the Laboratoires d'Excellence "Immunotherapy-Graft-Oncology" (LabEx IGO). RNA-seq was performed by the ICGex NGS platform of the Institut Curie, which is supported by grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche (“Investissements d’Avenir” Program), Multi-Organisation Thematic Institutes for Cancer (ITMO-CANCER), and the SIRIC-Curie Program (SIRIC grant INCa-DGOS- 4654). Proteomic studies were supported by Région Ile-de-France and Fondation pour la Recherche Médicale grants (D.L.). Some BM plasma was obtained from patients included in the BIO-FLIRT (Follicular Lymphoma IV/SC Rituximab Therapy) study as a part of the ancillary study of the FLIRT trial (NCT02303119) sponsored by the Lymphoma Academic Research Organisation. The immunofluorescence study was performed on the Microscopy Rennes Imaging Center (MRic-ALMF; UMS 6480 Biosit, Rennes, France). Transmission electron microscopy was done at the same platform microscopy center in collaboration with Agnes Burel.
Authorship
Contribution: E.D. designed and performed experiments, analyzed data, and wrote the paper; C.P. provided data on FL B-cell GEP; D.R. and S.L. contributed to data analysis; M.D. and C.C. provided the SMAD2/3-SMAD4 reporter cell line; T.M. contributed to study design; M.L. and J.D. provided technical assistance; P.L. ran RNA sequencing; F.D. carried out the MS experimental work; D.L. supervised MS experiments and data analysis; E.F. and G.C. provided samples; T.F. provided samples and contributed to study design; and K.T. designed and supervised research and wrote the paper.
Conflict-of-interest disclosure: This study used some samples collected using Bio-FLIRT funding as a part of the ancillary FLIRT study. The authors declare no other competing financial interests.
Correspondence: Karin Tarte, INSERM, UMR 1236, Faculté de Médecine, 2 Ave du Pr Léon Bernard, 35043 Rennes, France; e-mail: karin.tarte@univ-rennes1.fr.