

Key Points
YBX1 is selectively required for maintaining survival of myeloid leukemia cells, but is dispensable for normal hematopoiesis.
YBX1 cooperates with IGF2BPs via its cold-shock domain and stabilizes the mRNA of BCL2 and MYC in an m6A-dependent manner.

Abstract
RNA-binding proteins (RBPs) are critical regulators of transcription and translation that are often dysregulated in cancer. Although RBPs are increasingly recognized as being important for normal hematopoiesis and for hematologic malignancies as oncogenes or tumor suppressors, RBPs that are essential for the maintenance and survival of leukemia remain elusive. Here we show that YBX1 is specifically required for maintaining myeloid leukemia cell survival in an N6-methyladenosine (m6A)-dependent manner. We found that expression of YBX1 is significantly upregulated in myeloid leukemia cells, and deletion of YBX1 dramatically induces apoptosis and promotes differentiation coupled with reduced proliferation and impaired leukemic capacity of primary human and mouse acute myeloid leukemia cells in vitro and in vivo. Loss of YBX1 has no obvious effect on normal hematopoiesis. Mechanistically, YBX1 interacts with insulin-like growth factor 2 messenger RNA (mRNA)-binding proteins (IGF2BPs) and stabilizes m6A-tagged RNA. Moreover, YBX1 deficiency dysregulates the expression of apoptosis-related genes and promotes mRNA decay of MYC and BCL2 in an m6A-dependent manner, which contributes to the defective survival that results from deletion of YBX1. Thus, our findings have uncovered a selective and critical role of YBX1 in maintaining myeloid leukemia survival, which might provide a rationale for the therapeutic targeting of YBX1 in myeloid leukemia.
Introduction
Eukaryotic cells regulate messenger RNA (mRNA) expression through transcriptional and posttranscriptional mechanisms. In these pro-cesses, RNA-binding proteins (RBPs) play essential roles by fine-tuning gene expression, RNA splicing, polyadenylation, stability, localization, translation, and degradation.1 RBPs typically bind RNA via 1 or several RNA-binding domains (RBDs) such as the RNA recognition motif, DEAD box domain, or K-homology domain.1,2 Recent advances uncovered additional RBPs that lack conventional RBDs.3 Alterations of RBPs have been implicated in different human cancers; thus, it is necessary to decipher the complicated regulatory mechanisms of RBPs and their cancer-related RNA targets, which will provide a better understanding of cancer biology and unveil potential therapeutic targets.4
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy characterized by uncontrolled expansion of poorly differentiated myeloid cells.5 Accumulated evidence reveals that RBPs are essential for normal hematopoiesis and hematopoietic malignancies.6-8 For instance, frequent mutations in RBPs that serve as splicing factors have been identified in myeloid dysplasia.9,10 N6-methyladenosine (m6A) is a modification commonly found in mammalian mRNA,11 and it plays a critical role in determining the fate of RNA.12,13 This modification is reversible and is catalyzed by a methyltransferase complex (METTL3-METTL14-WTAP) and demethylases ALKBH5 and FTO.14-16 Several key m6A regulators have emerged as critical players in normal and malignant hematopoiesis.17-26 Our recent studies27,28 reveal that the RNA m6A demethylase ALKBH5 is selectively required for maintaining the function of AML leukemic stem cells but not normal hematopoietic stem cells (HSCs). Despite these recent advances, it is still necessary to explore RBPs that are essential to leukemia maintenance and survival, which will contribute to understanding the pathogenesis of AML development.
YBX1 belongs to the RBP family and is a multifunctional protein that contains the evolutionarily conserved cold-shock domain (CSD). YBX1 has been implicated in various biological pro-cesses because it affects a wide range of genes involved in cell proliferation, survival, drug resistance, and chromatin destabilization.29,30 YBX1 can also act as a versatile oncoprotein and is required for tumor cell proliferation, progression, and metastasis.31,32 However, the role of YBX1 in leukemia remains elusive. Here, we show that YBX1 regulates m6A-tagged mRNA stability by interacting with insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) and is selectively required for maintaining the survival of myeloid leukemia cells.
Methods
Mice
C57BL/6J (CD45.2) background Ybx1fl/fl and NOD.Cg-PrkdcscidIL2rgnull (B-NSG) mice were obtained from Biocytogen. Mx1-cre mice and B6.SJL (CD45.1) were purchased from The Jackson Laboratory. All of the animal experiments were carried out according to the protocols approved by Animal Care and Use Committee of the Medical Research Institute, Wuhan University.
Primary samples from patients with AML
Patients with AML provided informed consent for collecting samples from bone marrow (BM) aspirations. All experiments that refer to human samples were conducted in compliance with all relevant ethical regulations and were approved by the ethics committees of medical research units of the various universities. Experimental details are provided in the supplemental Data, available at the Blood Web site.
Generation of the murine MLL-AF9 leukemia model
BM cells were extracted from 8- to 12-week-old Ybx1cKO and wild-type (WT) mice, and lineage-negative cells (Lin–) were enriched and used for generating MLL-AF9 AML mice. Experimental details are provided in the supplemental Data.
Functional, biochemical, and molecular assays
Colony formation assay, competitive repopulation assays, cell proliferation, cell cycle differentiation and apoptosis analyses, cell staining, and flow cytometry analyses were performed. We performed RNA pull-down, RNA immunoprecipitation (RIP), protein expression and purification, western blotting, co-immunoprecipitation, RNA decay, luciferase reporter assays, RIP-polymerase chain reaction (RIP-PCR), quantitative reverse transcriptase-PCR (qRT-PCR), methylated RIP-PCR (MeRIP-PCR), RNA sequencing (RNA-seq), m6A sequencing (m6A-seq), and thiol(SH)-linked alkylation for the metabolic sequencing of RNA (SLAM-seq). Experimental details are provided in the supplemental Data.
Results
High expression of YBX1 in human myeloid leukemia cells
To comprehensively investigate the expression of RBPs in AML, we first analyzed the expression levels of 1068 RBP mRNAs in patients with AML by surveying the publicly available data sets from The Cancer Genome Atlas (TCGA). Interestingly, we found that YBX1 had higher expression in patients with AML and ranked among the top candidates in all RBPs (Figure 1A). Moreover, YBX1 mRNA expression in human AML samples was significantly higher than that in other cancer types (Figure 1B). We also found that YBX1 was expressed at a significantly higher level in leukemia cells from de novo patients with AML, includ-ing patients with normal karyotype, inv(16), MLL-rearranged t(11q23), and t(8,21), than in healthy controls (Figure 1C). And YBX1 was also aberrantly elevated at the protein level in samples from AML patients compared with those from healthy control cells (Figure 1D). Similar higher YBX1 protein levels were detected in various human leukemia cell lines (Figure 1E). Together, these data suggest that YBX1 might play a critical role in myeloid leukemia.

Elevated expression of YBX1 is required for survival of human myeloid leukemia cells. (A) Expression profiling of 1068 RNA-binding protein-encoding genes in patients with AML using the TCGA database. (B) YBX1 mRNA had the highest level of expression in AML compared with other cancers among 1429 human cancer cell lines in the Cancer Cell Line Encyclopedia database (https://portals.broadinstitute.org/ccle). Horizontal bars represent the tumor types where 1429 cells lines belong. (C) qRT-PCR analysis was used to determine the expression of YBX1 in normal BM cells from healthy donors (n = 5) and patient-derived primary AML cells of various subtypes, including inv(16) (n = 8), normal karyotype (n = 23), t(11q23) (n = 13), and t(8;21) (n = 28). Horizontal bars represent the cells used for qPCR are derived from bone marrow cells from healthy samples and patient samples with different karyotypes. (D) Immunoblot showing YBX1 expression in bulk BM mononuclear cells from healthy donors (n = 2) and primary patients with AML (n = 5). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as a loading control. (E) Western blot analysis of YBX1 expression in various patient-derived myeloid leukemia cell lines; CD34+ cells derived from cord blood were used as a normal control and GAPDH served as the loading control. (F) Immunoblot showing YBX1 KD efficiency in leukemia cells, MOLM-13, and THP-1 after transduction with shRNA lentiviruses targeting YBX1. (G) Colony formation assay showing the clonogenic defect of primary Lin–CD34+ cells from 3 individual patients with AML after YBX1 KD. Horizontal bars (AML#171, AML#172, and AML#173) are names of 3 patient samples. (H) In vivo leukemic engraftment analysis at 8 weeks after xenotransplantation. Data from 3 independent experiments for 3 patients with AML were combined (right, 3 recipients per patient sample per group). Horizontal bars (AML#XH04, AML#ZN01, and AML#124) are names of 3 patient samples. (I) Growth curves of MOLM-13 and THP-1 leukemia cells after transduction with lentiviruses for shYBX1#1, shYBX1#2, or shControl. (J) Colony formation assay of MOLM-13 and THP-1 leukemia cells after transduction with indicated lentiviruses. Horizontal bars (YBX1-KD#1 and YBX1-KD#2) represent shYBX1#1 and shYBX1#2, respectively. (K) Growth curves showing the rescued growth of leukemia cells by ectopic expression of YBX1. Leukemia cells were transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ UTR plus YBX1 complementary DNA for rescuing YBX1 expression. (L) Colony formation assay showing the rescued clonogenic capability of leukemia cells by ectopic expression of YBX1. Horizontal bars represent leukemia cells transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ UTR plus YBX1 complementary DNA for rescuing YBX1 expression or shControl. (M) Cell cycle distribution of leukemia cells after transduction with the indicated lentiviruses. Cells were labeled with Hoechst 33342 and assessed by flow cytometry. Horizontal bars represent different phases of the cell cycle after leukemia cells were transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ plus YBX1 complementary DNA for rescuing YBX1 expression or shControl. (N) Percentages of apoptotic leukemia cells at day 4 after transduction. Horizontal bars represent leukemia cells transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ UTR plus YBX1 complementary DNA for rescuing YBX1 expression or shControl. (O) Kaplan-Meier plot showing the survival time of 3 cohorts of recipient mice transplanted with MOLM-13 cells (n = 4 to 5 recipient mice per group). Horizontal bars represent days after leukemia cells were transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ UTR plus YBX1 complementary DNA for rescuing YBX1 expression or shControl and injected to recipient mice. A log-rank test was performed, and panels F to N show 1 representative sample of 3 independent experiments. Error bars denote mean ± standard deviation (SD). Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. FPKM, fragments per kilobase million; GFP, green fluorescent protein.
Elevated expression of YBX1 is required for survival of human myeloid leukemia cells. (A) Expression profiling of 1068 RNA-binding protein-encoding genes in patients with AML using the TCGA database. (B) YBX1 mRNA had the highest level of expression in AML compared with other cancers among 1429 human cancer cell lines in the Cancer Cell Line Encyclopedia database (https://portals.broadinstitute.org/ccle). Horizontal bars represent the tumor types where 1429 cells lines belong. (C) qRT-PCR analysis was used to determine the expression of YBX1 in normal BM cells from healthy donors (n = 5) and patient-derived primary AML cells of various subtypes, including inv(16) (n = 8), normal karyotype (n = 23), t(11q23) (n = 13), and t(8;21) (n = 28). Horizontal bars represent the cells used for qPCR are derived from bone marrow cells from healthy samples and patient samples with different karyotypes. (D) Immunoblot showing YBX1 expression in bulk BM mononuclear cells from healthy donors (n = 2) and primary patients with AML (n = 5). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as a loading control. (E) Western blot analysis of YBX1 expression in various patient-derived myeloid leukemia cell lines; CD34+ cells derived from cord blood were used as a normal control and GAPDH served as the loading control. (F) Immunoblot showing YBX1 KD efficiency in leukemia cells, MOLM-13, and THP-1 after transduction with shRNA lentiviruses targeting YBX1. (G) Colony formation assay showing the clonogenic defect of primary Lin–CD34+ cells from 3 individual patients with AML after YBX1 KD. Horizontal bars (AML#171, AML#172, and AML#173) are names of 3 patient samples. (H) In vivo leukemic engraftment analysis at 8 weeks after xenotransplantation. Data from 3 independent experiments for 3 patients with AML were combined (right, 3 recipients per patient sample per group). Horizontal bars (AML#XH04, AML#ZN01, and AML#124) are names of 3 patient samples. (I) Growth curves of MOLM-13 and THP-1 leukemia cells after transduction with lentiviruses for shYBX1#1, shYBX1#2, or shControl. (J) Colony formation assay of MOLM-13 and THP-1 leukemia cells after transduction with indicated lentiviruses. Horizontal bars (YBX1-KD#1 and YBX1-KD#2) represent shYBX1#1 and shYBX1#2, respectively. (K) Growth curves showing the rescued growth of leukemia cells by ectopic expression of YBX1. Leukemia cells were transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ UTR plus YBX1 complementary DNA for rescuing YBX1 expression. (L) Colony formation assay showing the rescued clonogenic capability of leukemia cells by ectopic expression of YBX1. Horizontal bars represent leukemia cells transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ UTR plus YBX1 complementary DNA for rescuing YBX1 expression or shControl. (M) Cell cycle distribution of leukemia cells after transduction with the indicated lentiviruses. Cells were labeled with Hoechst 33342 and assessed by flow cytometry. Horizontal bars represent different phases of the cell cycle after leukemia cells were transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ plus YBX1 complementary DNA for rescuing YBX1 expression or shControl. (N) Percentages of apoptotic leukemia cells at day 4 after transduction. Horizontal bars represent leukemia cells transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ UTR plus YBX1 complementary DNA for rescuing YBX1 expression or shControl. (O) Kaplan-Meier plot showing the survival time of 3 cohorts of recipient mice transplanted with MOLM-13 cells (n = 4 to 5 recipient mice per group). Horizontal bars represent days after leukemia cells were transduced with shRNA lentivirus targeting the YBX1 3′ UTR (shYBX1-3′ UTR) or shYBX1 3′ UTR plus YBX1 complementary DNA for rescuing YBX1 expression or shControl and injected to recipient mice. A log-rank test was performed, and panels F to N show 1 representative sample of 3 independent experiments. Error bars denote mean ± standard deviation (SD). Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. FPKM, fragments per kilobase million; GFP, green fluorescent protein.
YBX1 is required for survival of human myeloid leukemia cells
To test the functional role of YBX1 in leukemic cells, we first knocked down YBX1 using 2 different short hairpin RNAs (shRNAs [shYBX1#1 and shYBX1#2]). Both shRNAs markedly deleted YBX1 expression at the protein and mRNA levels (Figure 1F; supplemental Figure 1A). We transduced Lin–CD34+ cells derived from patients with AML, which are commonly considered a leukemia stem and progenitor cell–enriched population, with lentiviral shYBX1. As expected, YBX1 knockdown (KD) significantly reduced clonogenic capacity and impaired the in vivo leukemia reconstitution ability of these cells (Figure 1G-H), indicating that YBX1 is required for maintaining the function of primary leukemia stem and progenitor cells derived from patients with AML. We next explored the role of YBX1 in human myeloid leukemia cell lines. YBX1 KD resulted in a substantial inhibition of cell proliferation and clonogenicity in both MOLM-13 and THP-1 AML cells (Figure 1I-J). In addition, YBX1 KD induced differentiation, moderately affected cell cycles, and significantly promoted apoptosis of leukemia cells that showed a higher percentage of annexin V+ cells and increased cleaved caspase 3 of leukemia cells upon deletion of YBX1 (supplemental Figure 1B-G).
To rule out an off-target possibility, we restored YBX1 expression by inserting its complementary DNA, which was resistant to shYBX1 3′ targeting of the 3′ untranslated region (UTR) (supplemental Figure 1H). As expected, restoration of YBX1 completely rescued the defects in cellular growth, clonogenic ability, cell cycle arrest, and apoptosis caused by YBX1 KD (Figure 1K-N), indicating that the inhibitory function of YBX1 deficiency was not caused by an shRNA off-target effect. In addition, YBX1 KD cells exhibited delayed leukemia development, which was also reversed by restoring the expression of WT YBX1 (Figure 1O). Together, these data indicate that YBX1 is required for maintaining survival of human myeloid leukemia cells.
Ybx1 is required for murine AML development
We next examined whether Ybx1 is required for murine leukemogenesis. We first knocked down Ybx1 in primary leukemia cells from MLL-AF9–induced AML mice (Figure 2A). The efficiency of Ybx1 KD was confirmed at the protein and mRNA levels (Figure 2B; supplemental Figure 2A). As expected, Ybx1 KD inhibited the proliferation and clonogenic capacity of leukemia cells (Figure 2C-E), moderately affected the cell cycle, and substantially induced apoptosis (Figure 2F-G; supplemental Figure 2B-C). Ybx1 KD leukemia cells were then transplanted into sublethally irradiated recipient mice. We found that recipients of Ybx1 KD leukemia cells developed AML and died as a result of AML significantly more slowly than did mice in the control group. This delayed AML development also correlated with a lower percentage of leukemia cells in peripheral blood (PB) and less severe splenomegaly (Figure 2H-J; supplemental Figure 2D-F). Together, these data indicate that Ybx1 is essential for murine AML development.

Ybx1 is required to maintain murine myeloid leukemia cell survival in vitro and in vivo. (A) Experimental scheme for assays in panels C to J. Lin– BM cells were transduced with MLL-AF9-yellow fluorescent protein (MLL-AF9-YFP) retrovirus and transplanted into sublethally irradiated recipient mice. YFP+ leukemia cells from AML mice were sorted and used for experiments as indicated. (B) Immunoblot showing Ybx1 KD efficiency in MLL-AF9-YFP leukemia cells after transduction with shRNA lentiviruses targeting Ybx1. (C) Growth curves of murine leukemia cells after transduction with lentiviruses for shYbx1#1, shYbx1#2, or shControl. (D-E) Colony formation assay (D; Control, shControl; P1 and P2, plating 1 and serial replating 2, respectively; Ybx1-KD#1 and Ybx1-KD#2, shYbx1#1 and shYbx1#2, respectively) and representative colony images (E; original magnification, ×100) of murine leukemia cells after transduction with the indicated lentiviruses; cells were cultured in MethoCult M3434 for 7 days. (F) Quantitative summary of flow cytometry analysis of cell cycles after leukemia cells were transduced with the indicated lentiviruses. Cells were labeled with Hoechst 33342 and were assessed by flow cytometry. (G) Percentage of apoptotic murine leukemia cells at day 4 after transduction. (H) Representative flow cytometry plot showing YFP+ leukemia cells in PB at 4 weeks after transplantation. (I) Percentages of YFP+ leukemia cells in PB at the indicated time after transplantation (n = 5 per group). (J) Kaplan-Meier survival curves of recipient mice transplanted with YFP+ leukemia cells after KD of Ybx1 (n = 5). A log-rank test was performed. (K) Experimental scheme for MLL-AF9–induced AML model. (L) Kaplan-Meier survival curves for recipients of MLL-AF9–transduced Lin– BM cells from WT or Ybx1cKO donor mice (n = 10; from 2 independent experiments). A log-rank test was performed. (M-N) Colony formation assay of leukemia cells (M) and cell numbers (N) from WT and Ybx1cKO AML mice. (B-G) Shown is 1 representative of ≥3 independent experiments. Error bars denote mean ± SD. Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. BMT, BM transplantation.
Ybx1 is required to maintain murine myeloid leukemia cell survival in vitro and in vivo. (A) Experimental scheme for assays in panels C to J. Lin– BM cells were transduced with MLL-AF9-yellow fluorescent protein (MLL-AF9-YFP) retrovirus and transplanted into sublethally irradiated recipient mice. YFP+ leukemia cells from AML mice were sorted and used for experiments as indicated. (B) Immunoblot showing Ybx1 KD efficiency in MLL-AF9-YFP leukemia cells after transduction with shRNA lentiviruses targeting Ybx1. (C) Growth curves of murine leukemia cells after transduction with lentiviruses for shYbx1#1, shYbx1#2, or shControl. (D-E) Colony formation assay (D; Control, shControl; P1 and P2, plating 1 and serial replating 2, respectively; Ybx1-KD#1 and Ybx1-KD#2, shYbx1#1 and shYbx1#2, respectively) and representative colony images (E; original magnification, ×100) of murine leukemia cells after transduction with the indicated lentiviruses; cells were cultured in MethoCult M3434 for 7 days. (F) Quantitative summary of flow cytometry analysis of cell cycles after leukemia cells were transduced with the indicated lentiviruses. Cells were labeled with Hoechst 33342 and were assessed by flow cytometry. (G) Percentage of apoptotic murine leukemia cells at day 4 after transduction. (H) Representative flow cytometry plot showing YFP+ leukemia cells in PB at 4 weeks after transplantation. (I) Percentages of YFP+ leukemia cells in PB at the indicated time after transplantation (n = 5 per group). (J) Kaplan-Meier survival curves of recipient mice transplanted with YFP+ leukemia cells after KD of Ybx1 (n = 5). A log-rank test was performed. (K) Experimental scheme for MLL-AF9–induced AML model. (L) Kaplan-Meier survival curves for recipients of MLL-AF9–transduced Lin– BM cells from WT or Ybx1cKO donor mice (n = 10; from 2 independent experiments). A log-rank test was performed. (M-N) Colony formation assay of leukemia cells (M) and cell numbers (N) from WT and Ybx1cKO AML mice. (B-G) Shown is 1 representative of ≥3 independent experiments. Error bars denote mean ± SD. Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. BMT, BM transplantation.
Next, we deleted Ybx1 in murine leukemia cells using CRISPR/Cas9-mediated knockout (KO) by 2 different guide RNAs (supplemental Figure 2G). Deletion of Ybx1 significantly reduced the clonogenic ability of leukemia cells (supplemental Figure 2H-I). We further generated Ybx1fl/fl mice and crossed them with the Mx1-Cre strain to get Mx1-cre;Ybx1fl/fl mice (supplemental Figure 3A). Eight- to 12-week-old Mx1-cre;Ybx1fl/fl and Ybx1fl/fl control mice were treated with polyinosinic-polycytidylic acid (pIpC) 3 times by intraperitoneal injection every other day. Efficient deletion of Ybx1 was observed at 2 and 4 weeks after the last injection of pIpC (supplemental Figure 3B-D). Because Mx1-Cre is also active in several major organs, we examined the histopathology 4 weeks after treatment with pIpC and did not detect any obvious defects in heart, liver, lung, kidney, or spleen tissue (supplemental Figure 3E). For simplicity, pIpC-treated Mx1-cre;Ybx1fl/fl mice will be referred to as Ybx1cKO mice, and pIpC-treated Ybx1fl/fl control mice will be referred to as WT mice. We found that loss of Ybx1 significantly delayed the development of AML, and recipients of MLL-AF9–transduced Ybx1cKO cells displayed markedly longer latency than did recipients of MLL-AF9–transduced WT Lin– cells (Figure 2K-L). This was also confirmed by a colony formation assay that showed fewer colonies in the absence of Ybx1 (Figure 2M-N). Taken together, our data indicate that Ybx1 is required for murine leukemogenesis.
Deletion of Ybx1 does not affect murine normal hematopoiesis
Next, we decided to determine whether YBX1 is required for normal hematopoiesis. We analyzed our in-house RNA-seq data sets (GSE165863) for 16 distinct hematopoietic populations from murine BM, but we did not find any significant difference in Ybx1 expression in these cells, which was further validated by qRT-PCR (supplemental Figure 3F-G). Compared with WT mice, Ybx1cKO mice showed comparable blood counts and frequencies of different types of blood cells in PB, including white blood cells, lymphoma cells, granulocytes, monocytes, platelets, and red blood cells, at 4 weeks after the last pIpC injection (Figure 3A-D; supplemental Figure 3H). The percentages and total numbers of long-term HSCs, short-term HSCs, multipotent progenitor population 2 (MPP2), MPP3, MPP4, Lin−Sca-1+c-Kit+(LSK) cells, lymphoid-primed multipotent progenitor (LMPP) cells, common myeloid progenitor (CMP), granulocyte/macrophage progenitor (GMP), megakaryocyte/erythroid progenitor (MEP), and common lymphoic progenitor (CLP) in the BM of Ybx1cKO mice were also similar to those in WT mice (Figure 3E-G; supplemental Figure 3I). Thus, these data indicate that Ybx1 is dispensable for normal hematopoiesis.
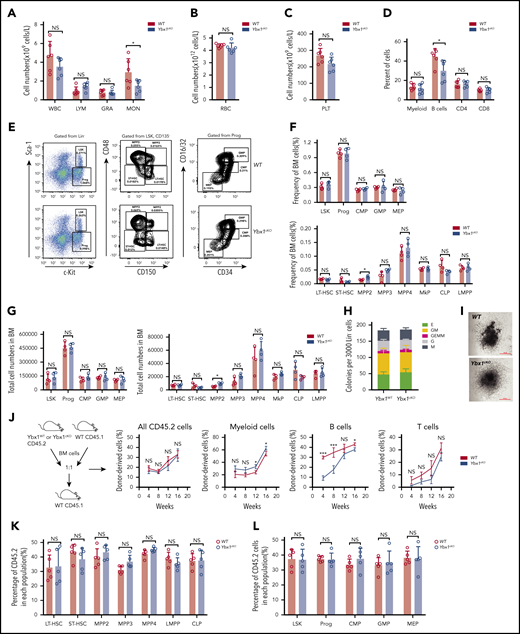
Ybx1 is dispensable for normal hematopoiesis. (A-C) Blood count analysis in the PB of WT and Ybx1cKO mice at 4 weeks after pIpC treatment (n = 6) for white blood cells (WBC), lymphoma cells (LYM), granulocytes (GRA), monocytes (MON) (A), red blood cells (RBC) (B), and platelets (PLT) (C). (D) Percentages of different mature lineage cells in the PB of WT and Ybx1cKO mice at 4 weeks after pIpC treatment (n = 6). (E) Representative fluorescence-activated cell sorting plots showing the gating strategy for different stem and progenitor cell populations in BM from WT and Ybx1cKO mice. (F-G)) Frequencies (F) and total cell numbers (G) of different progenitor populations (left) and stem cell populations (right) in the bone marrow of WT and Ybx1cKO mice 4 weeks after pIpC treatment (n = 4). (H-I) Colony formation assay showing similar clonogenic ability (H) and representative clone image (I) of Lin– BM cells from WT and Ybx1cKO mice at 4 weeks after pIpC treatment. (J-K) Competitive repopulation assay showing similar reconstitution capacity of HSCs from WT and Ybx1cKO mice. (J) Total BM cells from WT and Ybx1cKO mice were transplanted into lethally irradiated recipient mice (CD45.1) together with equal numbers of CD45.1 competitor BM cells. Flow cytometry analysis of different donor-derived cell lineages in PB of recipient mice from 4 to 16 weeks after BMT (WT, n = 5; Ybx1cKO, n = 5). (K) Percentage of donor-derived stem cell (left) and progenitor cell (right) compartments in the BM of recipients at 16 weeks after BMT. Horizontal bars represent abbreviations for cell types (A-D,F-G,K-L), or show the weeks after bone marrow transplantation (J). Error bars denote mean ± SD. Two-tailed Student t test: *P < .05; ***P < .001. LT-HSC, long-term HSC; MkP, megakaryocyte progenitor; NS, not significant; Prog, Lin−c-Kit+ progenitor cells; Sca-1, stem cell antigen-1; ST-HSC, short-term HSC.
Ybx1 is dispensable for normal hematopoiesis. (A-C) Blood count analysis in the PB of WT and Ybx1cKO mice at 4 weeks after pIpC treatment (n = 6) for white blood cells (WBC), lymphoma cells (LYM), granulocytes (GRA), monocytes (MON) (A), red blood cells (RBC) (B), and platelets (PLT) (C). (D) Percentages of different mature lineage cells in the PB of WT and Ybx1cKO mice at 4 weeks after pIpC treatment (n = 6). (E) Representative fluorescence-activated cell sorting plots showing the gating strategy for different stem and progenitor cell populations in BM from WT and Ybx1cKO mice. (F-G)) Frequencies (F) and total cell numbers (G) of different progenitor populations (left) and stem cell populations (right) in the bone marrow of WT and Ybx1cKO mice 4 weeks after pIpC treatment (n = 4). (H-I) Colony formation assay showing similar clonogenic ability (H) and representative clone image (I) of Lin– BM cells from WT and Ybx1cKO mice at 4 weeks after pIpC treatment. (J-K) Competitive repopulation assay showing similar reconstitution capacity of HSCs from WT and Ybx1cKO mice. (J) Total BM cells from WT and Ybx1cKO mice were transplanted into lethally irradiated recipient mice (CD45.1) together with equal numbers of CD45.1 competitor BM cells. Flow cytometry analysis of different donor-derived cell lineages in PB of recipient mice from 4 to 16 weeks after BMT (WT, n = 5; Ybx1cKO, n = 5). (K) Percentage of donor-derived stem cell (left) and progenitor cell (right) compartments in the BM of recipients at 16 weeks after BMT. Horizontal bars represent abbreviations for cell types (A-D,F-G,K-L), or show the weeks after bone marrow transplantation (J). Error bars denote mean ± SD. Two-tailed Student t test: *P < .05; ***P < .001. LT-HSC, long-term HSC; MkP, megakaryocyte progenitor; NS, not significant; Prog, Lin−c-Kit+ progenitor cells; Sca-1, stem cell antigen-1; ST-HSC, short-term HSC.
We next investigated the impact of Ybx1 deletion on HSC function. A colony formation assay showed normal clonogenic ability and differentiation potential of Ybx1cKO stem and progenitor cells (Figure 3H-I). To determine whether Ybx1 affects the self-renewal capacity of HSCs, we performed a competitive repopulation assay. We found that the percentages of WT and Ybx1cKO donor-derived total cells (CD45.2+), myeloid cells (Mac1+, Gr-1+), and T cells (CD3e+) in PB were comparable over 4 to 16 weeks after BM transplantation (Figure 3J). Interestingly, Ybx1 deficiency impaired B-cell reconstitution (Figure 3J), which needs to be studied in the future. At 16 weeks, we did not observe significant differences in the percentages of donor-derived WT and Ybx1cKO stem and progenitor cells (long-term HSCs, short-term HSCs, MPP2, MPP3, MPP4, LSK, LMPP, progenitor cells, CMP, GMP, MEP, and CLP) (Figure 3K-L; supplemental Figure 3J-K). Collectively, these results indicate that loss of Ybx1 does not affect normal hematopoiesis.
YBX1 stabilizes m6A-tagged RNA by cooperating with IGF2BPs
YBX1 has been shown to bind and stabilize RNA.33 To profile YBX1 binding RNA at the transcriptome level, we overexpressed FLAG-YBX1 in HEK293T cells and performed RIP after high-throughput sequencing (RIP-seq). We identified 9728 high-confidence binding sites within 2898 transcripts, which were highly enriched in promoter, exon, and 3′ UTRs (Figure 4A; supplemental Figure 4A). The majority of YBX1-binding RNAs (79.34%) were protein-encoding RNAs (supplemental Figure 4B). Gene ontology (GO) analysis showed that YBX1-binding transcripts were enriched in mRNA processing, splicing, and metabolic processes (supplemental Figure 4C). Surprisingly, we found that YBX1-binding sites clearly coincided with the m6A sites (Figure 4B; supplemental Figure 4D-E), which drove us to examine whether YBX1 binds to m6A-tagged RNAs. We conducted an RNA pull-down assay that used methylated single-stranded RNA bait (ss-m6A oligo with the consensus sequence GG(m6A)CU). Unmethylated control RNA (ss-A) was used as the control (Figure 4C). Interestingly, similar to positive control IGF2BPs, YBX1 from whole-cell nuclear lysate was pulled down by ss-m6A oligo but not ss-A oligo (Figure 4D), suggesting that YBX1 could bind to m6A-tagged RNAs. To assess whether YBX1 directly or indirectly binds to ss-m6A oligo, we purified glutathione S-transferase (GST)-YBX1 and used it in the RNA pull-down assay. Unlike IGF2BPs, purified YBX1 was not immunoprecipitated by ss-m6A (Figure 4E), implying that YBX1 indirectly binds to m6A-tagged RNAs.

YBX1 binds m6A-tagged RNA mediated by IGF2BPs. (A) The distribution of YBX1-binding peaks within different gene regions identified by FLAG-YBX1 RIP-seq in HEK293T cells. (B) Metagene profiles showing higher overlap of YBX1-binding sites and m6A modifications across the mRNA transcriptome. GSE29714 data were used for MeRIP-seq analysis. (C) Experimental scheme for RNA pull-down assay. Biotin-labeled single-strand RNA ss-m6A and biotin-labeled ss-A were used. (D) Western blot shows that IFG2BPs and YBX1 protein were selectively pulled down from HEK293T nuclear extract. (E) RNA pull-down assay showing the pull-down of purified GST-IGF2BP1/3 proteins, but not GST-YBX1 by biotin-labeled ss-m6A. (F) Coimmunoprecipitation and western blot showing the binding of YBX1 with IGF2BP1/3 in HEK293T cells. (G) Schematic structures showing the organization of YBX1 protein and YBX1 variants used in this study. Green box, the ala (A) and pro (P) enrich domain (A/P-rich domain); yellow box, an evolutionarily conserved CSD; pink and white separated boxes showing long C-terminal domain (CTD) with 4 arginine-rich motifs (ARMs) (pink boxes) and 4 acid motifs (AcidMs) (white boxes). (H-I) Coimmunoprecipitation and western blot showing the binding of FLAG-tagged YBX1 and its variants with hemagglutinin antigen (HA)-tagged IGF2BP1 in HEK293T cells (H) and of FLAG-CSD with endogenous IGF2BP1/3 in HEK293T cells (I). (J) RNA pull down with ss-m6A or ss-A in YBX1 KD or control HEK293T cells. Western blotting showing the similar pull-down efficiency of IGF2BP1/3 between YBX1 KD and control group. (K) RNA pull down with ss-m6A or ss-A in IGF2BPs KD or control HEK293T cells. Western blots showing the reduced pull-down amounts of IGF2BP1/3 and YBX1 between IGF2BP1/3 KD and control group. (L) Venn diagram showing the percentages of shared high-confidence targets among YBX1 and IGF2BPs. IGF2BPs iCLIP-seq data from GSE90686 were compared with YBX1 RIP-seq data. (M) Top consensus sequences of YBX1-binding sites, IGF2BP-binding sites, and the m6A motif detected by motif analysis. (N) YBX1 RIP-qPCR showing binding of FLAG-YBX1 to the representative targets (MYC, BCL2, TK1, MARCKSL1) in HEK293T cells. (O) Reducing mRNA half-life of the representative targets (MYC, BCL2, TK1, MARCKSL1) by deleting YBX1 in HEK293T cells. (P) Relative Fluc activity of MYC and BCL2 3′ UTR reporters with wild-type (CRD-WT) or mutated (CRD-mut) CRD in HEK293T cells with overexpressed YBX1. (Q) Relative luciferase activity (Rluc) ofCRD-WT orCRD-mut in IGF2BP1 KDor controlHEK293T cellswith expression of YBX1. (R) Relative luciferase activity ofCRD-WT or CRD-mut in YBX1 KD or control HEK293T cells with expression of IGF2BP1. RNA pull-down assays and western blotting show 1 representative of 3 independent experiments (D-K). Panels N and P-R show 1 representative of at least 3 independent experiments. Two-tailed Student t test: **P < .01; ***P < .001.; IP, immunoprecipittion; mut, mutant; shCon, shControl; Vec, vector.
YBX1 binds m6A-tagged RNA mediated by IGF2BPs. (A) The distribution of YBX1-binding peaks within different gene regions identified by FLAG-YBX1 RIP-seq in HEK293T cells. (B) Metagene profiles showing higher overlap of YBX1-binding sites and m6A modifications across the mRNA transcriptome. GSE29714 data were used for MeRIP-seq analysis. (C) Experimental scheme for RNA pull-down assay. Biotin-labeled single-strand RNA ss-m6A and biotin-labeled ss-A were used. (D) Western blot shows that IFG2BPs and YBX1 protein were selectively pulled down from HEK293T nuclear extract. (E) RNA pull-down assay showing the pull-down of purified GST-IGF2BP1/3 proteins, but not GST-YBX1 by biotin-labeled ss-m6A. (F) Coimmunoprecipitation and western blot showing the binding of YBX1 with IGF2BP1/3 in HEK293T cells. (G) Schematic structures showing the organization of YBX1 protein and YBX1 variants used in this study. Green box, the ala (A) and pro (P) enrich domain (A/P-rich domain); yellow box, an evolutionarily conserved CSD; pink and white separated boxes showing long C-terminal domain (CTD) with 4 arginine-rich motifs (ARMs) (pink boxes) and 4 acid motifs (AcidMs) (white boxes). (H-I) Coimmunoprecipitation and western blot showing the binding of FLAG-tagged YBX1 and its variants with hemagglutinin antigen (HA)-tagged IGF2BP1 in HEK293T cells (H) and of FLAG-CSD with endogenous IGF2BP1/3 in HEK293T cells (I). (J) RNA pull down with ss-m6A or ss-A in YBX1 KD or control HEK293T cells. Western blotting showing the similar pull-down efficiency of IGF2BP1/3 between YBX1 KD and control group. (K) RNA pull down with ss-m6A or ss-A in IGF2BPs KD or control HEK293T cells. Western blots showing the reduced pull-down amounts of IGF2BP1/3 and YBX1 between IGF2BP1/3 KD and control group. (L) Venn diagram showing the percentages of shared high-confidence targets among YBX1 and IGF2BPs. IGF2BPs iCLIP-seq data from GSE90686 were compared with YBX1 RIP-seq data. (M) Top consensus sequences of YBX1-binding sites, IGF2BP-binding sites, and the m6A motif detected by motif analysis. (N) YBX1 RIP-qPCR showing binding of FLAG-YBX1 to the representative targets (MYC, BCL2, TK1, MARCKSL1) in HEK293T cells. (O) Reducing mRNA half-life of the representative targets (MYC, BCL2, TK1, MARCKSL1) by deleting YBX1 in HEK293T cells. (P) Relative Fluc activity of MYC and BCL2 3′ UTR reporters with wild-type (CRD-WT) or mutated (CRD-mut) CRD in HEK293T cells with overexpressed YBX1. (Q) Relative luciferase activity (Rluc) ofCRD-WT orCRD-mut in IGF2BP1 KDor controlHEK293T cellswith expression of YBX1. (R) Relative luciferase activity ofCRD-WT or CRD-mut in YBX1 KD or control HEK293T cells with expression of IGF2BP1. RNA pull-down assays and western blotting show 1 representative of 3 independent experiments (D-K). Panels N and P-R show 1 representative of at least 3 independent experiments. Two-tailed Student t test: **P < .01; ***P < .001.; IP, immunoprecipittion; mut, mutant; shCon, shControl; Vec, vector.
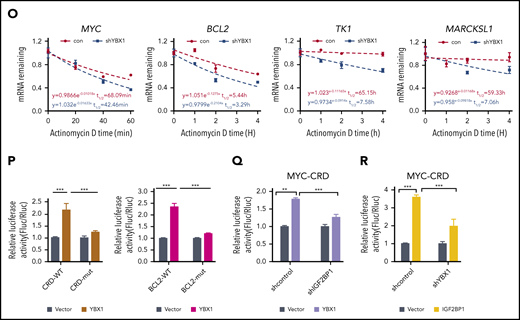
In line with a previous study,34 we found that YBX1 interacted with insulin-like growth factor 2 mRNA-binding proteins 1, 2, and 3 (IGF2BP1/2/3) (supplemental Figure 4F). Interactions between endogenous YBX1 and IGF2BP1/3 were observed (Figure 4F), and we also found that the YBX1 CSD mediated the interaction with IGF2BPs (Figure 4G-I; supplemental Figure 4G). Given that IGF2BPs are m6A readers, we proposed that IGF2BPs mediate the binding of YBX1 with m6A-tagged RNAs. To confirm our hypothesis, we knocked down IGF2BPs. An RNA pull-down assay showed that IGF2BP1/3 KD significantly abolished the binding of YBX1 with ss-m6A, whereas the YBX1 KD did not affect the binding of IGF2BPs with ss-m6A (Figure 4J-K). Thus, these data indicate that IGF2BPs are essential in mediating the binding of YBX1 to m6A-tagged RNAs. We further integrated our analysis with data from MeRIP-seq and RIP-seq for IGF2BPs and YBX1. Interestingly, ∼72.18% to 84.95% of the YBX1-bound transcripts were also bound by IGF2BPs (Figure 4L; supplemental Figure 4H), and both YBX1 and IGF2BPs preferentially bind the classic m6A motif containing the UGGAC consensus sequence (Figure 4M). As shown, representative high-confidence targets of IGF2BPs,35 including MYC, MARCKLS1, and TK1, were also enriched by YBX1 (Figure 4N; supplemental Figure 4I). Interestingly, we observed strong binding of the BCL2 transcript with YBX1 (Figure 4N). Functionally, we found that decay of MYC, BCL2, MARCKLS1, and TK1 mRNA were accelerated upon KD of YBX1 in HEK293T cells (Figure 4O). Thus, these results indicate that YBX1 regulates m6A-tagged mRNA stability, which is mediated by IGF2BPs.
To determine whether the effect of YBX1 on mRNA stability is m6A dependent, we chose to study MYC and BCL2. The firefly luciferase (Fluc) reporter that was inserted into the 249-bp WT or the m6A site’s mutant coding region instability determinant (CRD) of the 3′ terminus of the MYC coding region was used.35 We also constructed Fluc reporters with an ∼258-bp BCL2 3′ UTR that is enriched with m6A modifications or m6A site mutants. As expected, ectopic expression of YBX1 induced a significant increase in Fluc activity of the WT MYC and BCL2 reporters. Conversely, this increase was largely impaired by mutations in the m6A consensus sites (Figure 4P). Consistent with a previous study,35 as a positive control, the relative Fluc mRNA level of CRD-wt, but not CDR-mutant (CRD-mut), was increased by IGF2BP1 or IGF2BP3 overexpression (supplemental Figure 4J). Moreover, deletion of IGF2BP1 significantly abolished the increase of luciferase activity induced by YBX1 (Figure 4Q), suggesting that IGF2BPs are required for mediating the role of YBX1 on mRNA stability. Next, we knocked down YBX1 to assess whether YBX1 is necessary for the functioning of IGF2BPs. We found that YBX1 KD caused a substantial reduction of luciferase activity induced by IGF2BP1/3 (Figure 4R; supplemental Figure 4K), indicating that YBX1 is also required for IGF2BPs in maintaining m6A-tagged mRNA stability.
We next performed crosslinking and immunoprecipitation sequencing (CLIP-seq) for YBX1 in AML cells and compared it with m6A-seq data for these cells. We consistently found that ∼57.7% of YBX1-bound transcripts were m6A tagged, and ∼78.17% of YBX1-bound targets were bound by IGF2BPs (supplemental Figure 5A-B). To further assess whether YBX1 maintains AML in an m6A-dependent manner, we abolished the binding of YBX1 with IGF2BPs. Because it is currently not possible to identify the exact site of YBX1 that binds to IGF2BPs, we focused on the YBX1 CSD because it mediates the interaction with IGF2BPs (Figure 4G-I). Compared with intact YBX1, restoration of YBX1 with deleted CSD (ΔCSD) failed to rescue the phenotypes of YBX1-deficient leukemia cells (supplemental Figure 5C-I), suggesting that CSD is required for YBX1 function in AML cells. In addition, YBX1 can function as a reader of RNA 5-methylcytosine (m5C) and stabilize mRNA through the indole ring of W65 in its CSD.36,37 However, the YBX1-W65A mutant successfully rescued the phenotypes of YBX1-deficient leukemia cells, suggesting that the function of YBX1 as an m5C reader is not required for its effect on myeloid leukemia. Meanwhile, YBX1-W65A, but not ΔCSD, restored the expression of BCL2 and MYC (supplemental Figure 5J-K). Collectively, our data demonstrate that YBX1 fine-tunes m6A-tagged RNA stability by cooperating with IGF2BPs.
Loss of YBX1 regulates apoptotic genes in an m6A-dependent manner in leukemia cells
To better understand the role of YBX1 in leukemogenesis, we sorted leukemia-initiating cells from WT and Ybx1cKO AML mice and compared their gene expression profiles using RNA-seq (Figure 5A). A total of 3631 genes (1578 downregulated and 2053 upregulated) was found to be significantly changed due to Ybx1 loss. GO analysis showed that these genes were enriched in mRNA processing, RNA splicing regulation, myeloid cell differentiation, and regulation of the intrinsic apoptotic signaling pathway (Figure 5B; supplemental Figure 6A). Gene set enrichment analysis also showed the enrichment of genes involved in unfolded protein response and Wilms tumor 1 (WT1)-associated protein (WTAP) downregulated targets in Ybx1cKO cells (supplemental Figure 6B-D). Consistent with increased apoptosis of leukemia cells due to YBX1 loss, deletion of Ybx1 downregulated prosurvival genes including Myc, Bcl2l1, Mcl1, Tmbim6, and Birc6 (Figure 5C). In contrast, expression of proapoptotic genes such as Bax, Dapk3, and Siva1 was substantially increased in Ybx1cKO leukemia cells (Figure 5D). Similar changes in apoptotic-related genes were also observed in human myeloid leukemia cells upon YBX1 KD (supplemental Figure 6E). Thus, these results indicate that YBX1 plays an important role in regulating the expression of apoptotic genes in myeloid leukemia cells.
![Loss of YBX1 regulates apoptotic genes in an m6A manner in leukemia cells. (A) Volcano map showing differential expression of Ybx1 targets in WT and Ybx1cKO leukemia-initiating cells (LICs). (B) GO enrichment analysis of terms enriched in differential pathways with significantly downregulated genes in Ybx1cKO LICs. qRT-PCR validation of the effect of Ybx1 on the expression levels of (C) Myc, Lgals3, Bcl2l1, P4hb, Rps3, Birc6, Aatf, Tmbim6, Mcl1, Wtap, and Rbm7 and (D) Mxd4, Sival, Phf10, Dynll2, Dapk3, Bag1, Vapb, and Bax in WT and Ybx1cKO leukemia cells. (E) Distribution pattern for m6A peaks in LICs from AML mice as determined by using m6A-seq. The distribution (density) across 5′ UTRs, CDS, and 3′ UTRs is identified by an m6A site plot. The m6A DRACH (D, A/G/U; R, A or G; H, A/C/U) motif was enriched (inset) (P = 10−38). (F) Percentages of various RNA species with m6A modification for MeRIP-seq of leukemia cells from AML mice. (G) An integrative genomics viewer (IGV) tracks displaying higher enrichment of m6A peaks for Bcl2l1, Mcl1, and Wtap transcripts. (H) MeRIP-qPCR analysis of m6A enrichment of Bcl2l1, Mcl1, and Wtap. (I) SLAM-seq analysis showing the cumulative distributions of global transcript half-life (t1/2) in WT and Ybx1cKO LICs. (J) SLAM-seq analysis showing the comparable half-life of non-m6A and m6A-tagged transcripts in WT and Ybx1cKO LICs. (Kolmogorov-Smirnov [K-S] test, P = 5 × 10−7). (K) The mRNA half-life (t1/2) of Myc and Bcl2l1 mRNA as determined by SLAM-seq. Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. ATP, adenosine triphosphate; IgG, immunoglobulin G; ncRNA, non-coding RNA; rRNA, ribosome RNA, snoRNA, small nucleolar RNA; snRNA, small nuclear RNA.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/1/10.1182_blood.2020009676/6/m_bloodbld2020009676f5-1.png?Expires=1769079412&Signature=l6BG7HDkx9B9Usq~akwyAude2xOD0URk3f2qVgfA-TE~MeYyakNAOU82KMe7GJD0xTLOsYrI1NZNCc3pHFyCR4nLzZ50yXS4-CJPkTKK~PXYLgVSXaNURvfEHziyXWVk0k-Ne1BTE0IE7Xjlf0gFY-q3FLeJ-gEz5w3pDDov2fdFRWBu0XxN~mdx4~z~3N1MCRB~IBi6YvCZgsGjDzRaFAckNWzIjLTW5veQVtWEsS1~jqUafWd-MAD8NpRf-~j5KNk30xqWw4OIe-oKR9pHrMliYrL0RbLl1RQHJCjU2CsT3mLOleMT18QGA0Rv3rM6Ihw6Vt1vNv2X27OIXehMrQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Loss of YBX1 regulates apoptotic genes in an m6A manner in leukemia cells. (A) Volcano map showing differential expression of Ybx1 targets in WT and Ybx1cKO leukemia-initiating cells (LICs). (B) GO enrichment analysis of terms enriched in differential pathways with significantly downregulated genes in Ybx1cKO LICs. qRT-PCR validation of the effect of Ybx1 on the expression levels of (C) Myc, Lgals3, Bcl2l1, P4hb, Rps3, Birc6, Aatf, Tmbim6, Mcl1, Wtap, and Rbm7 and (D) Mxd4, Sival, Phf10, Dynll2, Dapk3, Bag1, Vapb, and Bax in WT and Ybx1cKO leukemia cells. (E) Distribution pattern for m6A peaks in LICs from AML mice as determined by using m6A-seq. The distribution (density) across 5′ UTRs, CDS, and 3′ UTRs is identified by an m6A site plot. The m6A DRACH (D, A/G/U; R, A or G; H, A/C/U) motif was enriched (inset) (P = 10−38). (F) Percentages of various RNA species with m6A modification for MeRIP-seq of leukemia cells from AML mice. (G) An integrative genomics viewer (IGV) tracks displaying higher enrichment of m6A peaks for Bcl2l1, Mcl1, and Wtap transcripts. (H) MeRIP-qPCR analysis of m6A enrichment of Bcl2l1, Mcl1, and Wtap. (I) SLAM-seq analysis showing the cumulative distributions of global transcript half-life (t1/2) in WT and Ybx1cKO LICs. (J) SLAM-seq analysis showing the comparable half-life of non-m6A and m6A-tagged transcripts in WT and Ybx1cKO LICs. (Kolmogorov-Smirnov [K-S] test, P = 5 × 10−7). (K) The mRNA half-life (t1/2) of Myc and Bcl2l1 mRNA as determined by SLAM-seq. Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. ATP, adenosine triphosphate; IgG, immunoglobulin G; ncRNA, non-coding RNA; rRNA, ribosome RNA, snoRNA, small nucleolar RNA; snRNA, small nuclear RNA.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/1/10.1182_blood.2020009676/6/m_bloodbld2020009676f5-2.png?Expires=1769079412&Signature=ZZ7Lyi~VDNmhUXDr~BGKybb923Qa-INXQor4UkYuLy1yCoCjrtQJuyqj3dvnWTV5APQc0XLb60hG11QRTLlkqVdinNUMeGZC4mFA94CZOgnvG5IXtChhedU5~3nSZuaNtwLWJ4~m7cFH5e2jNM9HsvQLn3O9SUrXV4YNXlYjPkOnxuFRwENoT1Law38iHczxZRfVpCXJmZUsOxyxIO6EUwLfMF1zTF13yldfWAXmPm~wvi8NblTt-WtWCuP~OSDGX~YXQ6YwWbDBLLGmwQwDLSc5TaCvUK2GPkf5h9lC0jONIhGFVUIFPhc~apCVkRYdr5z3bpn9WQQwjN6TjL9daA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Loss of YBX1 regulates apoptotic genes in an m6A manner in leukemia cells. (A) Volcano map showing differential expression of Ybx1 targets in WT and Ybx1cKO leukemia-initiating cells (LICs). (B) GO enrichment analysis of terms enriched in differential pathways with significantly downregulated genes in Ybx1cKO LICs. qRT-PCR validation of the effect of Ybx1 on the expression levels of (C) Myc, Lgals3, Bcl2l1, P4hb, Rps3, Birc6, Aatf, Tmbim6, Mcl1, Wtap, and Rbm7 and (D) Mxd4, Sival, Phf10, Dynll2, Dapk3, Bag1, Vapb, and Bax in WT and Ybx1cKO leukemia cells. (E) Distribution pattern for m6A peaks in LICs from AML mice as determined by using m6A-seq. The distribution (density) across 5′ UTRs, CDS, and 3′ UTRs is identified by an m6A site plot. The m6A DRACH (D, A/G/U; R, A or G; H, A/C/U) motif was enriched (inset) (P = 10−38). (F) Percentages of various RNA species with m6A modification for MeRIP-seq of leukemia cells from AML mice. (G) An integrative genomics viewer (IGV) tracks displaying higher enrichment of m6A peaks for Bcl2l1, Mcl1, and Wtap transcripts. (H) MeRIP-qPCR analysis of m6A enrichment of Bcl2l1, Mcl1, and Wtap. (I) SLAM-seq analysis showing the cumulative distributions of global transcript half-life (t1/2) in WT and Ybx1cKO LICs. (J) SLAM-seq analysis showing the comparable half-life of non-m6A and m6A-tagged transcripts in WT and Ybx1cKO LICs. (Kolmogorov-Smirnov [K-S] test, P = 5 × 10−7). (K) The mRNA half-life (t1/2) of Myc and Bcl2l1 mRNA as determined by SLAM-seq. Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. ATP, adenosine triphosphate; IgG, immunoglobulin G; ncRNA, non-coding RNA; rRNA, ribosome RNA, snoRNA, small nucleolar RNA; snRNA, small nuclear RNA.
Loss of YBX1 regulates apoptotic genes in an m6A manner in leukemia cells. (A) Volcano map showing differential expression of Ybx1 targets in WT and Ybx1cKO leukemia-initiating cells (LICs). (B) GO enrichment analysis of terms enriched in differential pathways with significantly downregulated genes in Ybx1cKO LICs. qRT-PCR validation of the effect of Ybx1 on the expression levels of (C) Myc, Lgals3, Bcl2l1, P4hb, Rps3, Birc6, Aatf, Tmbim6, Mcl1, Wtap, and Rbm7 and (D) Mxd4, Sival, Phf10, Dynll2, Dapk3, Bag1, Vapb, and Bax in WT and Ybx1cKO leukemia cells. (E) Distribution pattern for m6A peaks in LICs from AML mice as determined by using m6A-seq. The distribution (density) across 5′ UTRs, CDS, and 3′ UTRs is identified by an m6A site plot. The m6A DRACH (D, A/G/U; R, A or G; H, A/C/U) motif was enriched (inset) (P = 10−38). (F) Percentages of various RNA species with m6A modification for MeRIP-seq of leukemia cells from AML mice. (G) An integrative genomics viewer (IGV) tracks displaying higher enrichment of m6A peaks for Bcl2l1, Mcl1, and Wtap transcripts. (H) MeRIP-qPCR analysis of m6A enrichment of Bcl2l1, Mcl1, and Wtap. (I) SLAM-seq analysis showing the cumulative distributions of global transcript half-life (t1/2) in WT and Ybx1cKO LICs. (J) SLAM-seq analysis showing the comparable half-life of non-m6A and m6A-tagged transcripts in WT and Ybx1cKO LICs. (Kolmogorov-Smirnov [K-S] test, P = 5 × 10−7). (K) The mRNA half-life (t1/2) of Myc and Bcl2l1 mRNA as determined by SLAM-seq. Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. ATP, adenosine triphosphate; IgG, immunoglobulin G; ncRNA, non-coding RNA; rRNA, ribosome RNA, snoRNA, small nucleolar RNA; snRNA, small nuclear RNA.
Next, we explored how YBX1 regulates the expression of these genes. Considering our findings,s shown in Figure 4, we focused on m6A-mediated mRNA stability. As expected, YBX1 KD did not affect the global m6A level (supplemental Figure 6F). To identify transcripts that are methylated in leukemia-initiating cells from AML mice, we mapped the m6A methylome using m6A-seq, and identified 7547 m6A peaks that were primarily enriched in both exons and the 3′ UTR around the stop codon and displayed the classic DRACH (D, A/G/U; R, A or G; H, A/C/U) motif (Figure 5E). In addition, >87% of the m6A-tagged transcripts were protein encoding (Figure 5F). The integrative genomics viewer displayed high enrichment of m6A peaks in Bcl2l1 and Mcl1 (Figure 5G). Transcripts of Rbm7 and Zfp36l1 were also modified with m6A (supplemental Figure 6G-H). As an important subunit of m6A methyltransferase complex, WTAP is a novel oncogenic protein for leukemogenesis, and it regulates proliferation of acute myeloid leukemia cells.38 Interestingly, we observed higher m6A peaks in a Wtap transcript (Figure 5G), suggesting that Ybx1 might affect Wtap expression in leukemia cells. MeRIP-qPCR further validated these m6A modifications on these transcripts (Figure 5H).
Notably, m6A modification affects mRNA stability.39 So we measured the transcriptome-wide mRNA decay in leukemia-initiating cells from WT and Ybx1cKO AML mice using SLAM-seq.40 Interestingly, we observed a significant increase of mRNA decay at the global level in Ybx1cKO leukemia cells (KO, 94.7 minutes vs WT, 130.3 minutes) (Figure 5I). To test the contribution of m6A modification to RNA degradation, we integrated m6A-seq data with SLAM-seq data and compared the degradation rates of m6A-tagged RNAs with non–m6A-tagged RNAs. As expected, m6A-tagged transcripts in both WT and Ybx1cKO leukemia cells showed accelerated degradation when compared with non–m6A-tagged transcripts. Importantly, the loss of Ybx1 increased the degradation of m6A-tagged transcripts (Figure 5J). For instance, mRNA stabilities of Myc and Bcl2l1 were significantly decreased upon Ybx1 deletion (Figure 5K), but deletion of Ybx1 did not regulate the transcription and splicing of its targets in leukemia cells (supplemental Figure 6I-L). Taken together, our data indicate that Ybx1 regulates the expression of apoptotic genes in leukemia cells by affecting their mRNA stabilities.
MYC and BCL2 mediate the function of YBX1 in leukemia cells
We observed significant enrichment of MYC targets and genes of oxidative phosphorylation in YBX1high cases from 2 AML patient cohorts (Figure 6A-C). BCL2 is involved in oxidative phosphorylation of AML stem cells,41 so we focused on c-MYC and BCL2. Downregulation of Myc and Bcl2 in Ybx1cKO leukemia cells from AML mice was confirmed (Figure 6D). YBX1 KD in human leukemia MOLM-13 and THP-1 cells also caused marked reduction of MYC and BCL2 expression (Figure 6E-F; supplemental Figure 7A). We also observed an interaction of YBX1 with IGF2BP1/3 in leukemia cells (supplemental Figure 7B). We investigated whether YBX1 regulates BCL2 and MYC expression by modulating their mRNA stability in leukemia cells. Indeed, loss of Ybx1 caused an obvious decrease in the half-life of BCL2 and MYC mRNA (Figure 6G). Similar results were also found in human leukemia cells upon YBX1 KD (supplemental Figure 7C). From our m6A-seq data, we observed higher enrichment of m6A peaks in Myc and Bcl2 transcripts in leukemia cells, which was validated by MeRIP-qPCR (Figure 6H-I). YBX1 RIP-PCR showed that YBX1 directly binds BCL2 and MYC transcripts, which was impaired with knockdown of IGF2BPs (Figure 6J; supplemental Figure 7D). Therefore, our data indicate that YBX1 stabilizes BCL2 and MYC mRNA in an m6A-dependent manner.

MYC and BCL2 mediate the function of YBX1 in leukemia cells. (A-C) Gene set enrichment analysis plots showing enrichment of MYC targets and genes for oxidative phosphorylation in YBX1high vs YBX1low groups from GSE14468 and TCGA AML patient cohorts. (D) Immunoblot for Myc and Bcl2 expression in WT and Ybx1cKO leukemia cells (Gapdh was used as a loading control). (E-F) Immunoblot for MYC and BCL2 expression in MOLM-13 cells (E) and THP-1 cells (F) 4 days after transduction with the indicated lentiviruses (GAPDH was used as a loading control). (G) The mRNA half-life of Myc and Bcl2 in WT and Ybx1cKO leukemia cells. (H) IGV tracks that show the distribution of m6A peaks for Myc and Bcl2 transcripts. (I) MeRIP-qPCR analysis of m6A enrichment of Myc and Bcl2 in LICs. Primers 1, 2, and 3 are located around m6A sites. (J) YBX1 RIP-qPCR analysis showing YBX1 binding to MYC and BCL2 mRNA in shControl or shIGF2BP1 leukemia cells. (K) Colony formation assay of LICs from WT and Ybx1cKO AML mice. Ybx1cKO LICs were transduced with lentiviruses expressing Myc or Bcl2. (L) Representative images from a colony formation assay indicating lentiviruses. Original magnification, ×100. (M-N) Growth curve (M) and colony formation assay (N) of MOLM-13 cells after transduction with indicated lentiviruses. (O) Flow cytometry analysis of apoptosis after transduction with indicated lentiviruses at day 4. (P) Percentages of apoptotic leukemia cells after transduction at day 4. (Q) Working model showing the role of YBX1 in leukemia. Panels D-G and I-P show 1 representative of at least 3 independent experiments. Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. FDR, false discovery rate; NES, normalized enrichment score.
MYC and BCL2 mediate the function of YBX1 in leukemia cells. (A-C) Gene set enrichment analysis plots showing enrichment of MYC targets and genes for oxidative phosphorylation in YBX1high vs YBX1low groups from GSE14468 and TCGA AML patient cohorts. (D) Immunoblot for Myc and Bcl2 expression in WT and Ybx1cKO leukemia cells (Gapdh was used as a loading control). (E-F) Immunoblot for MYC and BCL2 expression in MOLM-13 cells (E) and THP-1 cells (F) 4 days after transduction with the indicated lentiviruses (GAPDH was used as a loading control). (G) The mRNA half-life of Myc and Bcl2 in WT and Ybx1cKO leukemia cells. (H) IGV tracks that show the distribution of m6A peaks for Myc and Bcl2 transcripts. (I) MeRIP-qPCR analysis of m6A enrichment of Myc and Bcl2 in LICs. Primers 1, 2, and 3 are located around m6A sites. (J) YBX1 RIP-qPCR analysis showing YBX1 binding to MYC and BCL2 mRNA in shControl or shIGF2BP1 leukemia cells. (K) Colony formation assay of LICs from WT and Ybx1cKO AML mice. Ybx1cKO LICs were transduced with lentiviruses expressing Myc or Bcl2. (L) Representative images from a colony formation assay indicating lentiviruses. Original magnification, ×100. (M-N) Growth curve (M) and colony formation assay (N) of MOLM-13 cells after transduction with indicated lentiviruses. (O) Flow cytometry analysis of apoptosis after transduction with indicated lentiviruses at day 4. (P) Percentages of apoptotic leukemia cells after transduction at day 4. (Q) Working model showing the role of YBX1 in leukemia. Panels D-G and I-P show 1 representative of at least 3 independent experiments. Two-tailed Student t test: *P < .05; **P < .01; ***P < .001. FDR, false discovery rate; NES, normalized enrichment score.
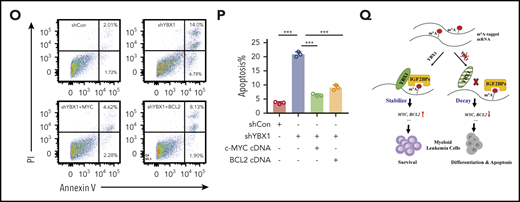
Next, we examined whether MYC and BCL2 mediate the function of YBX1 in leukemia cells. We re-expressed either MYC or BCL2 in primary mouse Ybx1cKO leukemia cells and YBX1 KD human leukemia cells, and we found that ectopic expression of MYC or BCL2 substantially rescued the clonogenic and proliferative defects resulting from YBX1 deficiency (Figure 6K-N; supplemental Figure 7E). Importantly, ectopic expression of MYC or BCL2 also prevented the induction of apoptosis and restored the cell cycle in YBX1 KD leukemia cells (Figure 6O-P; supplemental Figure 7F-G). Finally, by using the significantly downregulated targets in Ybx1cKO leukemia cells as a gene cluster, we surveyed their expression in AML patients from the TCGA database. We found that the expression levels of 420 mRNAs were positively correlated with YBX1 expression in patients with AML, and higher expression of these 420 signature genes correlated with their shorter overall survival (supplemental Figure 7H-J). Taken together, these data indicate that BCL2 and MYC are functional downstream targets of YBX1.
Discussion
Our study demonstrates that YBX1, as an RNA-binding protein, is selectively required for maintaining myeloid leukemia cell survival but is dispensable for normal hematopoiesis. This function of YBX1 is accomplished by stabilizing m6A-tagged mRNA (including BCL2 and MYC) by interacting with m6A reader IGF2BPs (Figure 6Q). Thus, our findings shed light on the underlying mechanism that controls survival of myeloid leukemia cells.
Although dysregulated expression of YBX1 has been observed in several human solid cancers,42-45 to our knowledge, this study provides the first clear evidence for the role of YBX1 in hematopoiesis and leukemogenesis. Surprisingly, we did not observe any significant changes in hematopoietic stem and progenitor cells in Ybx1cKO mice. Although a previous study using an EML cell line implied that YBX1 may function in the early stage of erythropoiesis,46 we did not detect any obvious defects in erythroid development, but we did show that deletion of YBX1 impairs leukemogenesis by inducing apoptosis and inhibiting proliferation of myeloid leukemia cells. A new study47 showed that YBX1 mediates persistence of JAK2-mutated myeloproliferative neoplasm cells, and the authors also found that deletion of YBX1 does not perturb normal hematopoiesis and the function of hematopoietic stem and progenitor cells. Therefore, our data have uncovered a unique and critical function of YBX1 in myeloid leukemia.
YBX1 is a multifunctional protein and is involved in regulating both transcription and translation.48 Surprisingly, our study revealed an important role of YBX1 in stabilizing m6A-tagged RNA by interacting with m6A reader IGF2BPs. We found that YBX1 is not an m6A reader, suggesting that YBX1 acts as either a stabilizer or a scaffold for recruiting other proteins. YBX1 also interacts with other m6A readers such as YTHDF2 and YTHDC1/2 (data not shown), suggesting that it might affect mRNA fates in multiple ways. Thus, the exact mechanism of how YBX1 works in regulating m6A-tagged RNA may be context dependent. In addition, YBX1 functions as a reader for another RNA modification, m5C, and maintains the stability of its target mRNA in the pathogenesis of bladder cancer and zebrafish embryogenesis.36,37 Given the multifaceted roles of YBX1 in regulating cotranscription and posttranscription, YBX1 may act in distinct manners on transcripts with different modifications. Thus, it will be important to further investigate the underlying mechanisms of YBX1 in different contexts.
Our findings establish a link between YBX1 and BCL2 expression. BCL2 is found to be upregulated in AML cells and leukemia stem cells,41,49 and inhibiting BCL2 via a selective BH3 mimetic such as venetoclax has proven to be an efficient and successful strategy in eradicating leukemic stem cells in clinics.50 Our study provides evidence that m6A modification is essential in maintaining the stability of BCL2 mRNA in myeloid leukemia cells, which is accomplished by YBX1. Modification of m6A could play multiple roles in determining BCL2 mRNA fate, and a recent study indicates that m6A promotes the translation of MYC and BCL2 mRNA in myeloid leukemia cells.18 Collectively, our results reveal novel mechanistic insights into how YBX1 selectively functions in regulating survival of myeloid leukemia cells and suggest a potential therapeutic strategy for treating myeloid leukemia.
Data can be found under accession number GSE159154.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
There is a Blood Commentary on this article in this issue.
Acknowledgments
The authors acknowledge the members of our laboratory for helpful discussion.
This work is supported by grants from the National Key Research and Development Program of China (2017YFA0505600) (H.Z.), the National Natural Science Foundation of China (81870124 and 81722003), the Wuhan Science and Technology Program for Application and Basic Research Project (2018060401011325), the Natural Science Foundation of Hubei Province (2019CFA073), the Hubei Provincial Natural Science Foundation for Creative Research Group (2018CFA018), and the Medical Science Advancement Program (Basic Medical Sciences) of Wuhan University (TFJC2018005). This work was also supported by a grant from the National Postdoctoral Fellowship (81500133) (J. Chang).
Authorship
Contribution: M.F., X.X., G.H., and H.Z. designed the experiments; M.F. and H.Z. contributed to the experimental plan and data interpretation; G.H. and Yicun Li performed bioinformatic analyses with help from J. Chang and Y.C. and with conceptual input from H.Z.; M.F. performed growth curve and colony formation assays, qPCR, and western blotting on primary samples or cell lines with help from Tong Zhang and Tiantian Zhang; G.H. performed mouse experiments with help from M.F., K.G., and M.C.; M.F. and Tiantian Zhang performed plasmid constructions with help from J.W. and P.W.; M.F. constructed the RNA-sequencing library; X.X. performed the SLAM-seq with help from M.F., R.Y., and Q.W.; Yashu Li constructed the m6A-sequencing library with help from J.H., Z.G., and C.G.; J. Chai and W.L. performed hematoxylin and eosin staining; S.L. and J. Chen helped with or advised on experiments; L.L. and F.Z. provided AML samples, clinical data, and materials; H.Z. and M.F. designed and supervised the overall study; and M.F., X.X., G.H., and H.Z. performed statistical analysis and wrote the manuscript.
Conflict-of-interest disclosure: J. Chen is a scientific founder of and holds equities with Genovel Biotech Corporation. The remaining authors declare no competing financial interests.
Correspondence: Haojian Zhang, Frontier Science Center for Immunology and Metabolism, Medical Research Institute, School of Medicine, Wuhan University, No.185 East Lake Rd, Wuchang District, Wuhan, Hubei 430071, People’s Republic of China; e-mail: haojian_zhang@whu.edu.cn.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal