As applications for machine learning in medicine continue to expand, demonstrate the utility of artificial intelligence algorithms for predicting prognosis in acute myeloid leukemia (AML) by defining novel molecular groupings.1
This is a fine example of a clinically actionable classification system based on machine learning that would not have been obvious to an experienced hematologist or in routine statistical methods. Machine learning is rapidly transforming medicine in radiology by automating the detection of clinically important patterns found within pixelated images,2 but its exact role in cancer management is still in its infancy.
AML is an excellent test bed for combinatorial biomarker testing in cancer: at least 50% of patients have a relatively stable karyotype, a low somatic mutation burden, and a high consistency of mutation recurrence.3-5 This means that the next new adult patient with de novo AML is likely to harbor at least 1 of the 22 most frequent somatic mutations, such as FLT3, NPM1, or TET2. Clinicians have known for more than a decade that mutations in combination matter. Patients with NPM1 mutation but without FLT3-ITD have a better prognosis than those with NPM1 and FLT3-ITD. Biallelic CEBPA-mutated AML has a better prognosis than one CEBPA mutation.6 But going beyond these examples and including more complex combinatorial mutation groupings is difficult and requires large sample sizes and computational algorithms. We also know from preleukemic stem cell studies that the acquisition order of mutation probably matters a great deal in AML (ie, whether you get a RUNX1 mutation first or a TET2 mutation first).7 Therefore, if we are to improve precision prognosis prediction, then we cannot consider genomic lesions in isolation.
The need for complementary prognosis classifiers is urgent: many normal karyotype patients, for instance, can simply not be categorized as high or low risk. These AMLs may lack biallelic CEBPA or FLT3-ITD but have a selection of somatic mutations (such as EZH2, IDH2, or GATA2) that, when considered in isolation, have not been shown to be particularly high risk or low risk. In this issue, Awada et al from the Maciejewski group at Cleveland Clinic elegantly demonstrate the power of machine-learning–type algorithms to reveal previously unrecognized subgroups of AML that are not obviously apparent. Their predictive model is available as a web-based open source resource (https://drmz.shinyapps.io/local_app/), although they emphasize that their proposed schema is complementary to rather than a replacement of current classifications.8
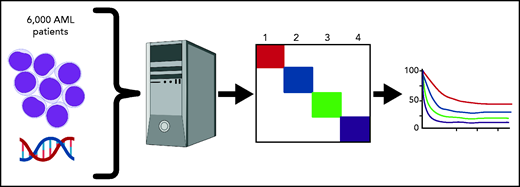
Awada et al chose to use a simple machine-learning algorithm, Bayesian latent class analysis, applied to 6788 patients for whom genomic data were available (see figure). Latent class analysis tries to fit mutations into 1 underlying “latent” disease type vs another until a classification is found in which no 2 diseases share the same associated features. The method is commonly used in analyzing data from questionnaires with yes/no responses or attributing symptoms to an underlying disease process. For instance, lack of smell plus fever plus dry cough is more likely to be COVID-19 than common rhinovirus based on Bayesian probability. Because it does assume any prior associations and does not take into account morphology or antecedent conditions (such as myelodysplasia), machine learning allowed the team to build a model of AML disease subtypes based on the probability of common molecular features belonging, or not, to a separate underlying “disease.”
Machine crunching was able to divide all patients into 4 major subgroups, which were then tested for prognostic value, that should show differences if indeed the new groupings represent distinct biology. The first (genomic cluster 1) had a median survival time of 34 months and comprised normal karyotype AML enriched for NPM1, DNMT3A, FLT3-ITD, and IDH2 R140 mutations but absent ASXL1, EZH2, TP53, and RUNX1. The second group (genomic cluster 2) had a median survival of 26 months and comprised biallelic CEBPA, IDH2 R172K with the absence of NPM1, ASXL1, RUNX1, and TP53. Cluster 3 (including SF3B1, SRSF2, and EZH2 mutations) and category 4 (abnormal karyotype) had median survival times of 15.8 months and 9 months, respectively. Importantly, cross-validation studies showed an accuracy of 0.97.
Interestingly, de novo and secondary AML did not separate out cleanly between the 4 clusters, alluding to the possibility that morphological criteria or prior history of a blood disorder may not be the best possible criteria for classification of this heterogenous disease. It is also noteworthy that IDH2 R140 mutations, but not IDH2 R172K or IDH1 mutations, were important classifiers for cluster 1, implying subtle differences in biology and metabolism. DNMT3A, IDH2 R140, and TET2 mutations were important determinants of cluster 2 but only if NPM1 mutation was not present, suggesting that the presence of the enigmatic NPM1 mutation dominates the phenotype. RUNX1 mutations were especially important in attributing membership to cluster 3 and held the second-highest global importance as a classifying marker, underscoring its inclusion as a provisional disease entity in the recent World Health Organization classification.9 Also of note, complex karyotype AML could be found in cluster 3 if the critical delineating lesions, −5/del(5q), −17/del(17p), and TP53 mutation, were not present, suggesting that some complex karyotype AML may not always be associated with worst outcome.
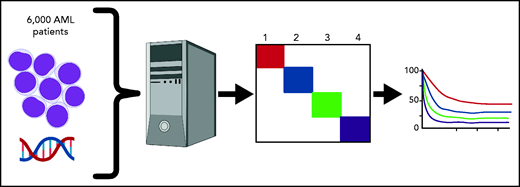
Genomic lesions including somatic mutations and translocations from >6000 AML patients were aggregated. Machine learning identified 4 underlying genomic clusters based on molecular features that were then shown to have differing survival rates, implying underlying differences in biology.
Genomic lesions including somatic mutations and translocations from >6000 AML patients were aggregated. Machine learning identified 4 underlying genomic clusters based on molecular features that were then shown to have differing survival rates, implying underlying differences in biology.
Future work will be needed to validate machine-learning groupings prospectively in other cohorts and to study the underlying biology of these potentially distinct molecular entities, including in the leukemia stem cell and at clonal hematopoiesis.10 Another limitation is the semi-arbitrary restraining of their model to just 4 groupings: other parameters or algorithms could likely build alternative groupings. No doubt we will see machine-learning classifications applied to many other cancer types in the near future, as well as a different set of classifiers for targeted therapies beyond “7 + 3” and allogeneic transplantation. The most obvious benefit will be for patients who do not have an obviously good or bad “bellwether” biomarker for clinical decision-making.
Conflict-of-interest disclosure: The author declares no competing financial interests.