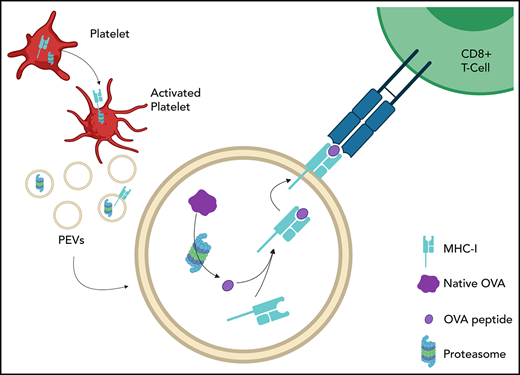
Key Points
Platelet-derived extracellular vesicles contain an active proteasome.
Platelet-derived extracellular vesicles can process and present antigen.

Abstract
In addition to their hemostatic role, platelets play a significant role in immunity. Once activated, platelets release extracellular vesicles (EVs) formed by the budding of their cytoplasmic membranes. Because of their heterogeneity, platelet EVs (PEVs) are thought to perform diverse functions. It is unknown, however, whether the proteasome is transferred from platelets to PEVs or whether its function is retained. We hypothesized that functional protein processing and antigen presentation machinery are transferred to PEVs by activated platelets. Using molecular and functional assays, we found that the active 20S proteasome was enriched in PEVs, along with major histocompatibility complex class I (MHC-I) and lymphocyte costimulatory molecules (CD40L and OX40L). Proteasome-containing PEVs were identified in healthy donor blood, but did not increase in platelet concentrates that caused adverse transfusion reactions. They were augmented, however, after immune complex injections in mice. The complete biodistribution of murine PEVs after injection into mice revealed that they principally reached lymphoid organs, such as spleen and lymph nodes, in addition to the bone marrow, and to a lesser extent, liver and lungs. The PEV proteasome processed exogenous ovalbumin (OVA) and loaded its antigenic peptide onto MHC-I molecules, which promoted OVA-specific CD8+ T-lymphocyte proliferation. These results suggest that PEVs contribute to adaptive immunity through cross-presentation of antigens and have privileged access to immune cells through the lymphatic system, a tissue location that is inaccessible to platelets.
Introduction
Platelets are the second most abundant lineage in the blood and are best known for their role in hemostasis.1 They are small fragments produced by the large, multinucleated megakaryocyte in the bone marrow. They bear receptors that permit recruitment of immune cells and carry an extensive set of immune and inflammatory molecules (eg, cytokines/chemokines, lipid mediators, and hormones) stored in their granules or cytoplasm or synthesized by messenger RNA translation after platelet activation. Thus, although platelets may mount an innate immune response against injury, which is critical to combating pathogen invasion, organ and tissue damage may also favor platelet activation and inflammation in chronic inflammatory diseases .2-8
Albeit anucleate, the platelet cytoplasm includes numerous molecules comprising the proteasome that are transferred from megakaryocytes to their progeny. The proteasome is a high-molecular-weight cylindrical protein complex through which unwanted or damaged proteins are degraded.9,10 The central complex part, called the 20S proteasome, is made up of 28 distinct subunits,11 comprising the 3 catalytic subunits necessary for the degradation of proteins into peptides of 3 to 15 amino acids in length.11,12 Proteasome activity in megakaryocytes is necessary for platelet production13,14 and in platelets, the proteasome regulates platelet lifespan,15 activation,16-18 and release of platelet EVs (PEVs).19,20 The platelet proteasome can hydrolyze proteins into smaller peptides,11,21,22 thereby enabling peptide loading onto the platelet major histocompatibility complex class I (MHC-I) molecules.23-25 Components of the peptide-loading complex are also expressed in platelets and are found in proximity with MHC-I during platelet activation.26,27 Platelets can efficiently form an immunological synapse with T lymphocytes to activate lymphocyte proliferation,26,28,29 and they are known to fulfill roles in cross-presentation of antigens in adaptive immunity. In a similar manner, megakaryocytes cross-present antigens to CD8 T lymphocytes, thereby suggesting that they may also play a dual role in innate and adaptive immunity.25,30-32
EVs, produced in abundance by platelets, are small (up to 1 µm in diameter), membrane-bound vesicles released from the plasma membrane or endosomal compartments of activated cells. PEVs are heterogeneous in surface molecules and content (eg, nucleic acids, lipids, transcription factors, enzymes, and mitochondria) and may have diverse functions beyond hemostasis.33-35 For instance, PEVs convey mitochondrial components that are associated with inflammation and adverse transfusion reactions (ATRs).36-38 Despite the fact that platelets are restricted to the blood circulation, PEVs can cross tissue barriers and enter the synovial fluid,39,40 lymph,41,42 and bone marrow43 where they can deliver platelet-derived molecules and modulate target cells.34 For instance, PEVs promote the formation of germinal centers and the production of IgG by B cells.44,45 They also interact with and modulate regulatory T-cell differentiation and activity.46,47 Thus, PEVs may be able to transport platelet-derived molecules relevant to adaptive immunity into lymphoid organs. However, it is unknown whether the proteasome and the molecules necessary for antigen presentation are also transferred during the budding of PEVs. In this study, we sought to find out whether functional protein processing and antigen presentation machinery are transferred to PEVs by activated platelets.
Material and methods
More details are presented in the supplemental Methods (available on the Blood Web site).
Labeling of murine platelets, DCs, and PEVs
Platelets were isolated from C57BL/6J mice by retro-orbital or cardiac puncture in 200 µL ACD and 350 µL Tyrode’s buffer (pH 6.5). Whole blood was centrifuged at 600g for 3 minutes and then at 400g for 2 minutes to remove red blood cells. Supernatant was spun at 1300g for 5 minutes, and the platelet-containing pellet was gently resuspended in 600 µL Tyrode’s buffer (pH 7.4). Platelets were either left nonactivated or activated with thrombin (0.1 U/mL) after addition of 5 mM of calcium for 90 minutes at room temperature (RT; time based on the kinetics of CD41+proteasome+ PEV release shown in supplemental Figure 3C). PEVs were obtained by 2 rounds of centrifugation of stimulated platelets at 1300g for 5 minutes at RT. Activated platelets, EVs, or dendritic cells (DCs) were pulsed with 100 µg/mL ovalbumin (OVA) protein (Sigma-Aldrich) or 200 µg/mL of OVA peptide (SIINFEKL [Invivogen]) or were left unpulsed for 4 hours at RT. These conditions were either left unlabeled for lymphoproliferation and intracellular staining experiments or labeled for Hs-flow cytometry (FCM) experiments.
Five microliters of PEVs or platelet suspensions were labeled with 250 nM LWA300 proteasome probe in a total volume of 100 μL for 90 minutes at 30°C. Samples were then incubated with the following antibodies for 30 minutes at RT before dilution in annexin V binding buffer and analysis by high sensitivity-FCM (hs-FCM): BUV395 anti-CD41, BV650 anti-CD62p, BUV395 anti-CD41, BV650 anti-CD62p, BV711 annexin V, BV421 anti-OX40L, BUV737 anti-CD154 (all from BD Biosciences), PeCy7 anti-CD40, PeCy7 anti-MHC-I (AF6-88.5), and PE anti-MHC-I bound to OVA peptide (25D1.16) (all from Biolegend).
Results
PEVs contain functional proteasome
After platelet activation by thrombin, remnant platelets were eliminated by centrifugation, and larger EVs were isolated by a second high-speed centrifugation (18 000g fraction). The supernatant obtained was further centrifuged at 100 000g, and smaller EVs (probably exosomes) were obtained from the pellet. We found that 98.1% ± 0.5% of proteins were retrieved in the larger EV (18 000g) fraction. Immunoblot analysis confirmed that human PEVs from this fraction were enriched in the proteasome 20S α-subunit, in addition to mitochondria (indicated by TOM20 expression) and CD41, but lacked TSG101 (a putative marker of exosomes; Figure 1A-E). Using platelets as a positive control, we assessed proteasome function in these PEVs. Proteasome-associated trypsinlike, caspaselike, and chymotrypsinlike activities were detectable in platelets and were significatively increased in the PEV fraction, but were undetectable after treatment with epoxomicin, a proteasome inhibitor (Figure 1F). Visualization of immunogold-labeled proteasome 20S α subunit by transmission electron microscopy confirmed the presence of proteasomes in PEVs (Figure 1G). These data suggest that the catalytically active proteasome was transferred to PEVs upon its release from the platelets.
![Platelets and PEVs contain proteasome. (A) Proteasome 20S α subunit, CD41, TOM20, TSG101 and actin in human PEVs (18 000g fraction) and platelet (PLTs) preparations (20 μg protein per lane) were assessed by immunoblot analysis. Results are representative of 5 distinct preparations. (B-E) Protein quantifications were assessed by densitometry with laboratory imaging software (BioRad). Results were normalized to actin and expressed as arbitrary units (AU). Mean ± SEM (n = 5). (D) *P < .05 (paired Student t test). (F) Proteasome function was assessed by measuring trypsin-like, caspase-like, or chymotrypsin-like activity of PEVs and platelets treated or not with epoxomicin using the Proteasome-Glo chymotrypsin-like, trypsin-like, and caspase-like cell-based assays. Twenty and 10 μg of proteins was used for platelets and PEVs, respectively. Mean ± SEM (n = 6). *P < .05, **P < .01, ***P < .001 (Mann-Whitney U test). (G) TEM visualization of immunogold labeling of proteasome 20S α subunit in PEVs released from thrombin (0.5 U/mL)-activated platelets (arrowheads). Data are representative of 3 independent experiments. (H) hs-FCM analysis of resting platelets and thrombin (0.5 U/mL)–activated platelets. Two distinct populations of PEVs, (larger PEVs [∼17% of these PEVs contain active proteasome] and smaller PEVs) that do not contain active proteasome (n = 20). Data are the mean ± SEM. **P < .01; ***P < .001; ****P < .0001 (Kruskal–Wallis). (I-J) Controls were performed to assess the specificity of PEV detection using hs-FCM. Sensitivity of CD41+proteasome+ PEVs to competition by epoxomicin, ultracentrifugation (Ultracentri), or 0.05% Triton X-100 and unlabeled samples are presented as the percentage of untreated (Control). Data are the mean ± SEM of 5 independent experiments. ****P < .0001 vs control (paired Student t test). (K) Confocal microscopy visualization of proteasome content associated with platelets (left) and PEVs (right). Visualization of CD41, wheat germ agglutinin (WGA) to determine plasma membrane surface and proteasome (LWA300), and merger of the stains is displayed in the region of interest (ROI). Populations originating from dashed-line squares and represented in ROI are triple positives (arrowheads) or CD41+ and WGA+ but proteasome− (arrows). SEM, standard error of the mean; TEM, transmission electron microscopy.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/25/10.1182_blood.2020009957/5/m_bloodbld2020009957f1.png?Expires=1769076000&Signature=yNYZNKn2-wUYfCxxB8vIMnj4lWGzhxof-R4zqJSETHFfOvrqxdjUkg8gcZonxp-1JkFuFxE~ZZZN~ClY4JFcZ9EfHLNF2egvmCTMCRFqJwp2FTSg5cf~JsvAooIY5m8cVufa2z8a65pZmTIup9EKuYQoWh3VifFkemicU2MS8EArCRXmm22AUjiYSxkruovf3~zwImRCIBIhvH7C1bo4~aBZVdWGuXDxJDybJ2zwasAV7HAEdv8oQQGhP6KSnqJ3AzAUqTSO4nSZkhr16kMRvn0zoHeZsjTtVac4NfOjWQ7L~jPb-oOz0fjRq7sVB35QF-QrvHz-kpLql6QTdrnQ1Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Platelets and PEVs contain proteasome. (A) Proteasome 20S α subunit, CD41, TOM20, TSG101 and actin in human PEVs (18 000g fraction) and platelet (PLTs) preparations (20 μg protein per lane) were assessed by immunoblot analysis. Results are representative of 5 distinct preparations. (B-E) Protein quantifications were assessed by densitometry with laboratory imaging software (BioRad). Results were normalized to actin and expressed as arbitrary units (AU). Mean ± SEM (n = 5). (D) *P < .05 (paired Student t test). (F) Proteasome function was assessed by measuring trypsin-like, caspase-like, or chymotrypsin-like activity of PEVs and platelets treated or not with epoxomicin using the Proteasome-Glo chymotrypsin-like, trypsin-like, and caspase-like cell-based assays. Twenty and 10 μg of proteins was used for platelets and PEVs, respectively. Mean ± SEM (n = 6). *P < .05, **P < .01, ***P < .001 (Mann-Whitney U test). (G) TEM visualization of immunogold labeling of proteasome 20S α subunit in PEVs released from thrombin (0.5 U/mL)-activated platelets (arrowheads). Data are representative of 3 independent experiments. (H) hs-FCM analysis of resting platelets and thrombin (0.5 U/mL)–activated platelets. Two distinct populations of PEVs, (larger PEVs [∼17% of these PEVs contain active proteasome] and smaller PEVs) that do not contain active proteasome (n = 20). Data are the mean ± SEM. **P < .01; ***P < .001; ****P < .0001 (Kruskal–Wallis). (I-J) Controls were performed to assess the specificity of PEV detection using hs-FCM. Sensitivity of CD41+proteasome+ PEVs to competition by epoxomicin, ultracentrifugation (Ultracentri), or 0.05% Triton X-100 and unlabeled samples are presented as the percentage of untreated (Control). Data are the mean ± SEM of 5 independent experiments. ****P < .0001 vs control (paired Student t test). (K) Confocal microscopy visualization of proteasome content associated with platelets (left) and PEVs (right). Visualization of CD41, wheat germ agglutinin (WGA) to determine plasma membrane surface and proteasome (LWA300), and merger of the stains is displayed in the region of interest (ROI). Populations originating from dashed-line squares and represented in ROI are triple positives (arrowheads) or CD41+ and WGA+ but proteasome− (arrows). SEM, standard error of the mean; TEM, transmission electron microscopy.
Platelets and PEVs contain proteasome. (A) Proteasome 20S α subunit, CD41, TOM20, TSG101 and actin in human PEVs (18 000g fraction) and platelet (PLTs) preparations (20 μg protein per lane) were assessed by immunoblot analysis. Results are representative of 5 distinct preparations. (B-E) Protein quantifications were assessed by densitometry with laboratory imaging software (BioRad). Results were normalized to actin and expressed as arbitrary units (AU). Mean ± SEM (n = 5). (D) *P < .05 (paired Student t test). (F) Proteasome function was assessed by measuring trypsin-like, caspase-like, or chymotrypsin-like activity of PEVs and platelets treated or not with epoxomicin using the Proteasome-Glo chymotrypsin-like, trypsin-like, and caspase-like cell-based assays. Twenty and 10 μg of proteins was used for platelets and PEVs, respectively. Mean ± SEM (n = 6). *P < .05, **P < .01, ***P < .001 (Mann-Whitney U test). (G) TEM visualization of immunogold labeling of proteasome 20S α subunit in PEVs released from thrombin (0.5 U/mL)-activated platelets (arrowheads). Data are representative of 3 independent experiments. (H) hs-FCM analysis of resting platelets and thrombin (0.5 U/mL)–activated platelets. Two distinct populations of PEVs, (larger PEVs [∼17% of these PEVs contain active proteasome] and smaller PEVs) that do not contain active proteasome (n = 20). Data are the mean ± SEM. **P < .01; ***P < .001; ****P < .0001 (Kruskal–Wallis). (I-J) Controls were performed to assess the specificity of PEV detection using hs-FCM. Sensitivity of CD41+proteasome+ PEVs to competition by epoxomicin, ultracentrifugation (Ultracentri), or 0.05% Triton X-100 and unlabeled samples are presented as the percentage of untreated (Control). Data are the mean ± SEM of 5 independent experiments. ****P < .0001 vs control (paired Student t test). (K) Confocal microscopy visualization of proteasome content associated with platelets (left) and PEVs (right). Visualization of CD41, wheat germ agglutinin (WGA) to determine plasma membrane surface and proteasome (LWA300), and merger of the stains is displayed in the region of interest (ROI). Populations originating from dashed-line squares and represented in ROI are triple positives (arrowheads) or CD41+ and WGA+ but proteasome− (arrows). SEM, standard error of the mean; TEM, transmission electron microscopy.
LWA300 is a conjugate between epoxomicin and BODIPY FL fluorophore that generates an activity-based, plasma membrane-permeable inhibitor that can identify the proteasome in cells.48,49 Using LWA300, we detected and quantified active proteasome-containing PEVs directly in the platelet secretome.48,49 hs-FCM confirmed PEV heterogeneity after platelet activation by thrombin (Figure 1H). Approximately 16.6% ± 6.5% of the larger (ie, ∼500-900 nm) PEVs50 contained proteasome, whereas the smaller vesicles (ie, <500 nm) had no detectable proteasome (Figure 1H; supplemental Figure 1). The detection specificity of proteasome-containing PEVs by hs-FCM was confirmed by using a combination of controls. We confirmed efficient competition of the LWA300 probe by unlabeled epoxomicin, and we determined the particulate nature and membrane moiety of proteasome-containing PEVs, as they were respectively pelleted by ultracentrifugation and sensitive to detergent treatment (Figure 1I-J). Confocal microscopic visualization of platelets as positive controls, and PEVs from thrombin-activated platelets labeled with LWA300 revealed that both platelets and a subpopulation of PEVs contained active proteasome (Figure 1K).
hs-FCM was further used to characterize proteasome-containing PEVs in terms of surface markers and mitochondrial content. Approximately half of the proteasome-containing PEVs exposed phosphatidylserine, whereas most expressed surface P-selectin (supplemental Figure 2A). Furthermore, 68.3% ± 7.8% of the proteasome-containing PEVs also contained mitochondria (supplemental Figure 2A). Investigation of the mechanisms underlying release of active proteasome+ PEVs revealed that the total number of PEVs (with and without proteasomes) was significantly reduced in the presence of actin inhibitors (cytochalasins B, D, and E and latrunculin A) but not by the tubulin polymerization inhibitor nocodazole (supplemental Figure 2B). Proteasome release in PEVs was not unique to thrombin stimulation, as ADP, cross-linked collagen–related peptide (CRP-XL), and heat-aggregated IgG (HA-IgG) also triggered release of proteasome-containing PEVs (supplemental Figure 2C).
Identification of proteasome-containing PEVs under physiological and pathological conditions
The presence of proteasome-containing PEVs was assessed under conditions conducive to platelet activation and PEV release. A mean of 1.82 × 106 (range, 1.13 × 105 to 8.11 × 106; n = 6) proteasome-containing PEVs per mL were detected by hs-FCM in the blood of healthy individuals, which corresponded to 2.6% ± 1.8% of the total PEVs in blood. PEVs were quantified in platelet concentrates (PCs) known to have caused ATRs and compared with control PCs that did not induce ATRs. Given the reported increase in mitochondria-containing PEVs in ATRs,36,37 we also determined their levels. High levels of proteasome-containing PEVs were found in all tested PCs (Figure 2A) but the concentrations of proteasome-containing PEVs (with or without mitochondria) were not significantly elevated in PCs that induced ATRs (Figure 2A). In contrast, compared with the controls, the concentrations of mitochondria-containing PEVs were increased in ATR-associated PCs, consistent with prior findings.36,37
![Identification of proteasome-containing PEVs under physiological and pathological conditions. (A) Proteasome-containing PEVs detected by hs-FCM are found in PFP from platelet concentrates that caused ATRs in recipients and in control concentrates that did not induce ATRs. The total number of proteasome-containing PEVs (containing mitochondria [mito] or not; proteasome+mito+ PEVs or proteasome+mito− PEVs) did not significantly differ between control and ATR, whereas proteasome−mito+ PEVs increased in ATR (no ATR group [n = 33] vs the ATR reaction group [n = 34], matched in duration of storage; data are the mean ± standard error of the mean [SEM]; ****P < .0001 (Student t test). NS, nonsignificant. (B) Proteasome-containing PEVs detected by hs-FCM are found in bronchoalveolar lavage fluid from mice after induction of transfusion-related acute lung injury (TRALI) with the 34-1-2s and AF6-88.5.5.3 antibodies and in control mice (n = 5) Data are the mean ± SEM; Student t test. NS, nonsignificant. (C) Proteasome-containing PEVs were detected at significantly higher levels in mice 1 hour after IV injection of HA-IgG vs control (diluent) mice (n = 3). Data are the mean ± SEM. **P< .01 (Student t test). PFP, platelet-free plasma.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/25/10.1182_blood.2020009957/5/m_bloodbld2020009957f2.png?Expires=1769076000&Signature=I9mViLn~o7pxqJaR3G~7-wugnkBk77HII9m8X6pOAvuQU1CnpuluBE3fbjrpxQWBjzFXNGqZUJz-Gy4KLj9neiragdRRJJji8pVEjwt21J4EQ0weg5xJ6v980exM8UYt7ENatWXM2PahgrCiotA7Cx4cLKKvAq4~vymLI5YiE-8dB~9eMXkfqFhH2MBfeMeX32cIprj6fG-Ie1jLlhR1DGN0IB~2cVvfiit6cY-OHDLunQkmgIkGQZm5UM~6gnu4gWEG08dn0QtjgmVrA5EsXQcj2am2oU6mAZ7uh7t3bIjDSEud9-v7WPUQgJ3J3dbPGGKijFlXrjnlr8ykZSGHpg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Identification of proteasome-containing PEVs under physiological and pathological conditions. (A) Proteasome-containing PEVs detected by hs-FCM are found in PFP from platelet concentrates that caused ATRs in recipients and in control concentrates that did not induce ATRs. The total number of proteasome-containing PEVs (containing mitochondria [mito] or not; proteasome+mito+ PEVs or proteasome+mito− PEVs) did not significantly differ between control and ATR, whereas proteasome−mito+ PEVs increased in ATR (no ATR group [n = 33] vs the ATR reaction group [n = 34], matched in duration of storage; data are the mean ± standard error of the mean [SEM]; ****P < .0001 (Student t test). NS, nonsignificant. (B) Proteasome-containing PEVs detected by hs-FCM are found in bronchoalveolar lavage fluid from mice after induction of transfusion-related acute lung injury (TRALI) with the 34-1-2s and AF6-88.5.5.3 antibodies and in control mice (n = 5) Data are the mean ± SEM; Student t test. NS, nonsignificant. (C) Proteasome-containing PEVs were detected at significantly higher levels in mice 1 hour after IV injection of HA-IgG vs control (diluent) mice (n = 3). Data are the mean ± SEM. **P< .01 (Student t test). PFP, platelet-free plasma.
Identification of proteasome-containing PEVs under physiological and pathological conditions. (A) Proteasome-containing PEVs detected by hs-FCM are found in PFP from platelet concentrates that caused ATRs in recipients and in control concentrates that did not induce ATRs. The total number of proteasome-containing PEVs (containing mitochondria [mito] or not; proteasome+mito+ PEVs or proteasome+mito− PEVs) did not significantly differ between control and ATR, whereas proteasome−mito+ PEVs increased in ATR (no ATR group [n = 33] vs the ATR reaction group [n = 34], matched in duration of storage; data are the mean ± standard error of the mean [SEM]; ****P < .0001 (Student t test). NS, nonsignificant. (B) Proteasome-containing PEVs detected by hs-FCM are found in bronchoalveolar lavage fluid from mice after induction of transfusion-related acute lung injury (TRALI) with the 34-1-2s and AF6-88.5.5.3 antibodies and in control mice (n = 5) Data are the mean ± SEM; Student t test. NS, nonsignificant. (C) Proteasome-containing PEVs were detected at significantly higher levels in mice 1 hour after IV injection of HA-IgG vs control (diluent) mice (n = 3). Data are the mean ± SEM. **P< .01 (Student t test). PFP, platelet-free plasma.
Transfusion-related acute lung injury (TRALI) is a potentially lethal adverse reaction that can result from transfusion of PCs.51 Thus, we quantified proteasome-containing PEVs in murine bronchoalveolar lavages in an inducible TRALI model.52,53 Proteasome-containing PEVs were detected in bronchoalveolar lavages from both TRALI and control mice (Figure 2B); however, no significant difference was observed between the 2 groups (Figure 2B), which suggests that proteasome-containing PEVs are not increased during lung inflammation in this model and therefore may not participate in the acute inflammation that characterizes the pathogenesis.
Our in vitro investigations pointed to the high potency of immune complexes (HA-IgG) in generating proteasome-containing PEVs (supplemental Figure 2C). Although mice lack FcγRIIA, that receptor is the only Fcγ receptor expressed by human platelets that is capable of responding to immune complexes.54 Recent findings indicate that circulating immune complexes stimulate the release of mitochondria-containing PEVs in mice expressing the FcγRIIA transgene.55,56 Compared with the diluent-injected control mice, the mice with immune-complex challenge showed significantly elevated levels of proteasome-containing PEVs in plasma (Figure 2C). These findings confirmed that proteasome-containing PEVs are present under various physiological and pathological conditions.
Protein processing by proteasome-containing PEVs
To study proteasome function in PEVs, we investigated its ability to process proteins into smaller peptides by assessing their successful loading into the antigen-binding groove of MHC-I molecules. We confirmed the expression of MHC-I on resting and thrombin-activated murine platelets and verified whether MHC-I is maintained on PEVs present in the platelet secretome. We found that washed resting platelets did not express MHC-I on their surface (Figure 3A-B); however, thrombin activation led to a significant increase in surface MHC-I expression (Figure 3A-B), consistent with the reported presence of this molecule in α-granules and its release upon activation.26,27,57,58 A small proportion (0.93% ± 0.13%) of the spontaneously released PEVs expressed MHC-I, but this proportion significantly increased upon platelet activation with thrombin (mean, 4.64% ± 0.98%).
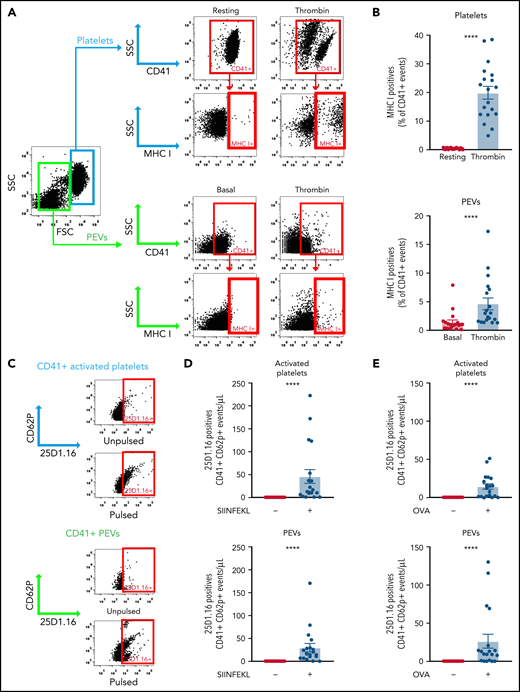
Platelets and PEVs load and process OVA onto MHC-I. (A-B) Thrombin (0.1 U/mL)–activated murine platelets and their PEVs express MHC-I (detected by hs-FCM; n = 19). Data are the mean ± standard error of the mean (SEM). ****P < .0001 (Mann-Whitney U test). Activated platelets and their PEVs loaded the SIINFEKL peptide (C-D) or processed and loaded OVA (C-E) onto MHC-I (n = 19). Data are the mean ± SEM. ****P < .0001, pulsed (+) vs unpulsed (−) (Kruskal-Wallis test).
Platelets and PEVs load and process OVA onto MHC-I. (A-B) Thrombin (0.1 U/mL)–activated murine platelets and their PEVs express MHC-I (detected by hs-FCM; n = 19). Data are the mean ± standard error of the mean (SEM). ****P < .0001 (Mann-Whitney U test). Activated platelets and their PEVs loaded the SIINFEKL peptide (C-D) or processed and loaded OVA (C-E) onto MHC-I (n = 19). Data are the mean ± SEM. ****P < .0001, pulsed (+) vs unpulsed (−) (Kruskal-Wallis test).
To determine whether PEV MHC-I can indeed load small peptides, we pulsed PEVs present in the platelet secretome with the OVA peptide SIINFEKL and monitored its association with MHC-I molecules using the 25D1.16 monoclonal antibody, which specifically recognizes MHC-I/SIINFEKL complexes.59 Similar to platelets, PEVs loaded the SIINFEKL peptide onto their MHC-I molecules (Figure 3C-D). Native OVA was also efficiently processed by platelets, and the SIINFEKL peptide was loaded into MHC-I (Figure 3C-E), consistent with prior work.26 We found that an average of 2.53% ± 0.74% of CD41+ PEVs pulsed with the peptide and 1.83% ± 0.24% of CD41+ PEVs pulsed with OVA (n = 18) were positive for 25D.1.16. Of interest, incubation of native OVA with PEVs resulted in proteolysis of the former and retrieval of the SIINFEKL peptide from MHC-I molecules expressed by the PEVs. Taken together, the data show that PEVs can process native proteins into smaller peptides thereby enabling antigen presentation through MHC-I.
Proteasome-containing PEVs can reach lymphoid organs and circulate through the lymphatic system
IV injected PEVs have a limited circulation time in human blood, ranging from 10 minutes to hours, depending on the study.60,61 It is unclear, however, whether they can reach lymphoid organs. Fluorescently labeled PEVs generated from activated mouse platelets were IV injected into mice, and their presence in blood and different organs was monitored. We identified free PEVs (unbound to cells) for up to 2 minutes in blood (Figure 4A; supplemental Figure 4A-C). PEVs in blood were also found bound to platelets and to leukocytes, mainly Ly6G+ neutrophils, and to a lesser extent, lymphocytes, but were mostly undetectable by 60 minutes (Figure 4A). Screening of individual PEVs in whole tissue sections in different organs identified spleen and lymph nodes (popliteal and inguinal) as primary targets, followed by liver, bone marrow, lungs, and kidneys, whereas none were found in brain (Figure 4B-C; supplemental Figure 4D-E). Moreover, aggregates of PEVs (ie, larger than 1 µm2 and up to 541 µm2) were mainly observed in spleen (mean size, 2.84 ± 0.16 µm2) and popliteal (4.16 ± 0.40 µm2) and inguinal (4.08 ± 0.30 µm2) lymph nodes, followed by bone marrow (2.75 ± 0.15 µm2), lung (33.51 ± 7.15 µm2), and liver (3.40 ± 0.21 µm2). This distribution may reflect their accumulation in smaller vessels or the internalization of numerous PEVs within single cellular recipients in these organs (Figure 4B-C; supplemental Figure 4D-E).
![PEVs in blood circulation can reach lymphoid organs and circulate in lymph. (A-C) Fluorescently labeled PEVs generated from activated mouse platelets were IV injected into mice and their presence in blood (A) and different organs (B-C) was monitored after 2, 15, and 60 minutes. Free PEVs (unbound to cells) were identified by flow cytometry for up to 2 minutes in blood, as well as PEVs bound to platelets and to leukocytes (mainly Ly6G+ neutrophils and a few lymphocytes), but were mostly undetectable by 60 minutes. Dashed lines represent the mean of vehicle (n = 9-13); n = 11 (2 minutes), n = 5 (15 minutes) and n = 6 (60 minutes). Data are the mean ± standard error of the mean (SEM). *P < .05, **P < .01, ***P < .001 (Kruskal-Wallis). (B) Representative images of CMFDA-labeled (green) individual PEVs (arrowhead) and PEV aggregates (asterisk) in whole tissue sections (spleen, popliteal LN [PLN], inguinal LN [ILN], bone marrow, lungs, and liver) at 15 and 60 minutes by confocal microscopy; nuclei (Hoechst 33342 ) are blue. Results are representative of observations made in 5 and 6 mice per group. (C) PEVs and aggregates were quantified using 5 different sections for lymph nodes (PLN and ILN; representing a total surface of at least 1.5 mm2), 8 zones of 500 000 µm2 each, randomly assigned on 2 different sections in femurs (total surface, 4 mm2), and 10 zones of 500 000 µm2 each, randomly assigned on 2 sections each of lungs, spleen, kidneys, and brain and 1 section of liver (total surface, 5 mm2) using Zen 3.3 software (n = 6 [phosphate buffered saline, 60 minutes], 5 [15 minutes], and 5-6 [60 minutes]). Data are the mean ± SEM. *P < .05, **P < .01 (Kruskal-Wallis). (D-E) PEVs in lymph were detected by hs-FCM. (D) Gating strategy to analyze expression of MHC-I and proteasome (LWA300) on CD41+ EVs in lymph and representative dot plot of labeled and unlabeled (CD41 only) lymph. (E) Expression of MHC-I and proteasome (LWA300) on CD41+ EVs in lymph was determined. +/+, double positive, and −/−, double negative for MHC-I and proteasome (n = 6). Data are the mean ± SEM.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/25/10.1182_blood.2020009957/5/m_bloodbld2020009957f4.png?Expires=1769076000&Signature=a4nqoSnaXyIHOjrnx1kjlydlP8MMprSFPkFUIH4yxLNTVZPndM~gVpDUrpbIVfojuczKO4lWUBzfaK0YU3yV6FiFoUhiVhu71O73FIzXj7vtCClZUXQQnnGhObuCh9Yox84GqrxNoH6qGbZwDWc3yIAfpf-D4E2iqyjuoeUVSujaA5Ff5rTNVW1Js67UUgreKCt-QdCeoXRgB7wjkncueQmMTlEovLvkYgsIaIX5JsSice7T-QeJ7~OBKA8PRy3xhJviEKFLb4eSMrD1l90fU2gvXDkk9y741Eoc2LRpOh4xeebt2apnRENnvEGUc6UvCcvUonXn0vawPx0YFYWEqw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PEVs in blood circulation can reach lymphoid organs and circulate in lymph. (A-C) Fluorescently labeled PEVs generated from activated mouse platelets were IV injected into mice and their presence in blood (A) and different organs (B-C) was monitored after 2, 15, and 60 minutes. Free PEVs (unbound to cells) were identified by flow cytometry for up to 2 minutes in blood, as well as PEVs bound to platelets and to leukocytes (mainly Ly6G+ neutrophils and a few lymphocytes), but were mostly undetectable by 60 minutes. Dashed lines represent the mean of vehicle (n = 9-13); n = 11 (2 minutes), n = 5 (15 minutes) and n = 6 (60 minutes). Data are the mean ± standard error of the mean (SEM). *P < .05, **P < .01, ***P < .001 (Kruskal-Wallis). (B) Representative images of CMFDA-labeled (green) individual PEVs (arrowhead) and PEV aggregates (asterisk) in whole tissue sections (spleen, popliteal LN [PLN], inguinal LN [ILN], bone marrow, lungs, and liver) at 15 and 60 minutes by confocal microscopy; nuclei (Hoechst 33342 ) are blue. Results are representative of observations made in 5 and 6 mice per group. (C) PEVs and aggregates were quantified using 5 different sections for lymph nodes (PLN and ILN; representing a total surface of at least 1.5 mm2), 8 zones of 500 000 µm2 each, randomly assigned on 2 different sections in femurs (total surface, 4 mm2), and 10 zones of 500 000 µm2 each, randomly assigned on 2 sections each of lungs, spleen, kidneys, and brain and 1 section of liver (total surface, 5 mm2) using Zen 3.3 software (n = 6 [phosphate buffered saline, 60 minutes], 5 [15 minutes], and 5-6 [60 minutes]). Data are the mean ± SEM. *P < .05, **P < .01 (Kruskal-Wallis). (D-E) PEVs in lymph were detected by hs-FCM. (D) Gating strategy to analyze expression of MHC-I and proteasome (LWA300) on CD41+ EVs in lymph and representative dot plot of labeled and unlabeled (CD41 only) lymph. (E) Expression of MHC-I and proteasome (LWA300) on CD41+ EVs in lymph was determined. +/+, double positive, and −/−, double negative for MHC-I and proteasome (n = 6). Data are the mean ± SEM.
PEVs in blood circulation can reach lymphoid organs and circulate in lymph. (A-C) Fluorescently labeled PEVs generated from activated mouse platelets were IV injected into mice and their presence in blood (A) and different organs (B-C) was monitored after 2, 15, and 60 minutes. Free PEVs (unbound to cells) were identified by flow cytometry for up to 2 minutes in blood, as well as PEVs bound to platelets and to leukocytes (mainly Ly6G+ neutrophils and a few lymphocytes), but were mostly undetectable by 60 minutes. Dashed lines represent the mean of vehicle (n = 9-13); n = 11 (2 minutes), n = 5 (15 minutes) and n = 6 (60 minutes). Data are the mean ± standard error of the mean (SEM). *P < .05, **P < .01, ***P < .001 (Kruskal-Wallis). (B) Representative images of CMFDA-labeled (green) individual PEVs (arrowhead) and PEV aggregates (asterisk) in whole tissue sections (spleen, popliteal LN [PLN], inguinal LN [ILN], bone marrow, lungs, and liver) at 15 and 60 minutes by confocal microscopy; nuclei (Hoechst 33342 ) are blue. Results are representative of observations made in 5 and 6 mice per group. (C) PEVs and aggregates were quantified using 5 different sections for lymph nodes (PLN and ILN; representing a total surface of at least 1.5 mm2), 8 zones of 500 000 µm2 each, randomly assigned on 2 different sections in femurs (total surface, 4 mm2), and 10 zones of 500 000 µm2 each, randomly assigned on 2 sections each of lungs, spleen, kidneys, and brain and 1 section of liver (total surface, 5 mm2) using Zen 3.3 software (n = 6 [phosphate buffered saline, 60 minutes], 5 [15 minutes], and 5-6 [60 minutes]). Data are the mean ± SEM. *P < .05, **P < .01 (Kruskal-Wallis). (D-E) PEVs in lymph were detected by hs-FCM. (D) Gating strategy to analyze expression of MHC-I and proteasome (LWA300) on CD41+ EVs in lymph and representative dot plot of labeled and unlabeled (CD41 only) lymph. (E) Expression of MHC-I and proteasome (LWA300) on CD41+ EVs in lymph was determined. +/+, double positive, and −/−, double negative for MHC-I and proteasome (n = 6). Data are the mean ± SEM.
PEVs can circulate through the lymphatic system, and the levels of PEVs in lymph are increased in mouse models of atherosclerosis and autoimmune inflammatory arthritis.34,41,42 Using the lymph from mice, we assessed to determine whether PEVs are associated with proteasome and MHC-I molecules. We found that a fraction of the PEVs in lymph expressed MHC-I (11.2% ± 2.2%) and contained an active proteasome (12.0% ± 3.9%). Remarkably, a detectable proportion (1.6% ± 0.7%) of the lymph PEVs contained both proteasome and MHC-I molecules (Figure 4D-E), significant given the substantial number of PEVs in lymph (mean, 2.5 × 107/mL in mice41).
Proteasome-containing PEVs express lymphocyte costimulatory molecules
Efficient stimulation of adaptive immunity requires both recognition of the antigen–MHC-I complexes by the T-cell receptor and the activity of costimulatory molecules. We examined to find whether platelets or proteasome-containing PEVs loaded with SIINFEKL express costimulatory molecules in addition to other known PEV markers displayed by CD41+proteasome+ EVs. Compared with PEVs that had undetectable SIINFEKL loading, both platelets and PEVs present in the platelet secretome loaded with SIINFEKL (25D1.16+) expressed higher levels of proteasome (Figure 5). Moreover, in contrast to thrombin-activated platelets, where phosphatidylserine expression is increased when loaded with SIINFEKL, both PEVs bearing SIINFEKL and those negative for SIINFEKL expressed similar levels of phosphatidylserine (Figure 5). Furthermore, both platelets and SIINFEKL-bearing PEVs expressed higher levels of P-selectin, and the costimulatory molecules CD40L, CD40, and OX40L (Figure 5). Thus, among the different subtypes of PEVs, those with a higher density of antigen-MHC-I complexes show more abundant expression of lymphocyte costimulatory molecules and bear a higher content of active proteasome.
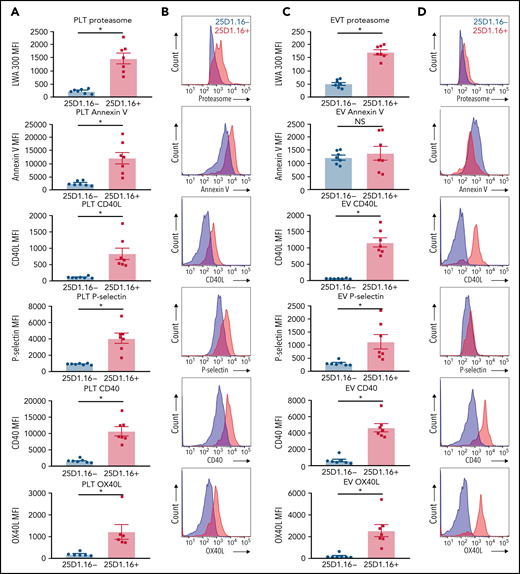
Platelets and PEVs loaded with OVA peptide express activation and costimulatory molecules. (A-B) Activated platelets and (C-D) PEVs loaded with OVA peptide (25D1.16+) express higher levels of proteasome (LWA300), activation (annexin V, P-selectin), and costimulatory molecules (CD40, CD40L, and OX40L). (A,C) Mean fluorescence intensity (MFI) of the different markers assessed by hs-FCM (n = 7). Data are mean ± standard error of the mean (SEM). *P < .05 (Student t test). NS nonsignificant. (B,D) Representative MFI histograms of the 25D1.16− and 25D1.16+ populations for each marker shown on CD41+proteasome+ events.
Platelets and PEVs loaded with OVA peptide express activation and costimulatory molecules. (A-B) Activated platelets and (C-D) PEVs loaded with OVA peptide (25D1.16+) express higher levels of proteasome (LWA300), activation (annexin V, P-selectin), and costimulatory molecules (CD40, CD40L, and OX40L). (A,C) Mean fluorescence intensity (MFI) of the different markers assessed by hs-FCM (n = 7). Data are mean ± standard error of the mean (SEM). *P < .05 (Student t test). NS nonsignificant. (B,D) Representative MFI histograms of the 25D1.16− and 25D1.16+ populations for each marker shown on CD41+proteasome+ events.
Proteasome-containing PEVs can support antigen-specific T-cell activation
T cells isolated from OT-1 mice were coincubated for 18 hours with PEVs present in the platelet secretome that were either pulsed or not with the SIINFEKL peptide or native OVA. DCs and platelets were treated similarly as positive controls and for comparison (Figure 6A). The T cells (CD3+CD8+) were then washed, and the expression of CD40, OX40, IL-2, and IFN-γ was recorded to assess T-cell activation.
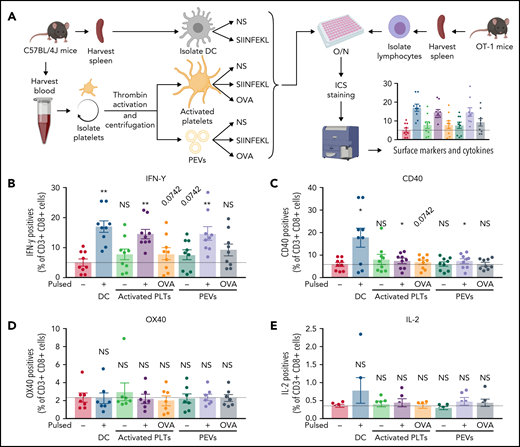
PEVs induce antigen-specific T-cell activation and cytokine production through antigen presentation. (A) The experimental plan. Cells and PEVs used for the stimulation of lymphocytes assessed by intracellular cytokines staining (ICS). NS, unpulsed; O/N, overnight. (B-E) Expression of receptors or cytokines by CD3+CD8+ T cells coincubated with DCs, activated platelets (PLTs), or PEVs left unpulsed or pulsed with SIINFEKL (PP), or OVA. IFN-γ production (B), CD40 expression (C), OX40 expression (D), and IL-2 production (E). Dashed lines are unstimulated conditions (n = 6, 7, or 9). Data are the mean ± SEM. *P < .05, **P < .01 vs unpulsed (Wilcoxon).
PEVs induce antigen-specific T-cell activation and cytokine production through antigen presentation. (A) The experimental plan. Cells and PEVs used for the stimulation of lymphocytes assessed by intracellular cytokines staining (ICS). NS, unpulsed; O/N, overnight. (B-E) Expression of receptors or cytokines by CD3+CD8+ T cells coincubated with DCs, activated platelets (PLTs), or PEVs left unpulsed or pulsed with SIINFEKL (PP), or OVA. IFN-γ production (B), CD40 expression (C), OX40 expression (D), and IL-2 production (E). Dashed lines are unstimulated conditions (n = 6, 7, or 9). Data are the mean ± SEM. *P < .05, **P < .01 vs unpulsed (Wilcoxon).
Compared with DCs and platelets, PEVs induced a significant release of IFN-γ when pulsed with the OVA peptide, whereas native OVA led to an increase in IFN-γ that did not reach statistical significance (Figure 6B). Moreover, DCs and, to a lesser extent, platelets and PEVs were capable of inducing only significant CD40 expression by T lymphocytes previously pulsed with the OVA peptide (Figure 6C). In contrast, OX40 and IL-2 expression was not induced by DCs, platelets, or PEVs under these experimental conditions (Figure 6D-E).
Whether PEVs could stimulate T-cell proliferation, a hallmark response by the lymphocyte antigen-MHC-I complex was explored. T cells from OT-1 mice were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) to monitor cellular division and were coincubated for 5 days with DCs, activated platelets, or PEVs, which were either pulsed or not with SIINFEKL or native OVA (Figure 7A). Lymphoproliferation is represented by a decrease in the mean fluorescence intensity histogram (ie, a dilution of CFSE fluorescence; Figure 7B).
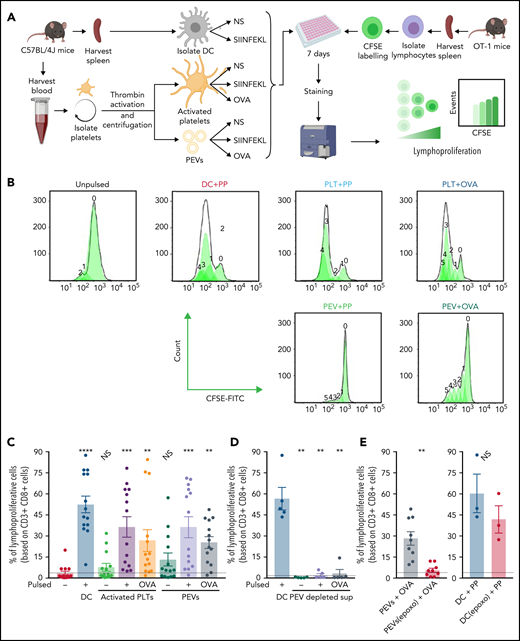
PEVs loaded with native OVA process and present OVA peptide to induce antigen-specific T-cell lymphoproliferation (A) The experimental plan. NS, unpulsed. (B) Histogram showing CFSE fluorescence shift of CD3+CD8+ T-cell populations when coincubated with DCs, activated platelets (PLTs), or PEVs left unpulsed or pulsed with SIINFEKL peptide (PP) or OVA for 7 days. (C) Percentage of CD3+CD8+ lymphoproliferative cells after coincubation with DCs, PLTs, or PEVs, unpulsed or pulsed with PP or OVA for 7 days. Data are the mean ± standard error of the mean (SEM); n = 14). **P < .01; ***P < .001; ****P < .0001 vs unpulsed (Friedman test followed by Dunn’s post hoc test for multiple comparisons). NS, nonsignificant. (D) Percentage of CD3+CD8+ lymphoproliferative cells after a 7-day coincubation with PP pulsed DCs or supernatant (surn), depleted of PEVs by ultracentrifugation, left unpulsed or pulsed with PP or OVA. Data are the mean ± SEM (n = 5). **P < .01 vs unpulsed (Mann-Whitney). (E) Proportion of CD3+CD8+ lymphoproliferative cells after a 7-day coincubation with OVA-pulsed PEVs, treated or not with epoxomicin (epoxo) for 2 hours, and PP-pulsed DCs (DC+PP), treated or not with epoxomicin (epoxo). Dashed lines are unstimulated conditions. Data are the mean ± SEM (n = 9 for PEVs and n = 3 for DC). **P < .01 (Wilcoxon). NS, nonsignificant.
PEVs loaded with native OVA process and present OVA peptide to induce antigen-specific T-cell lymphoproliferation (A) The experimental plan. NS, unpulsed. (B) Histogram showing CFSE fluorescence shift of CD3+CD8+ T-cell populations when coincubated with DCs, activated platelets (PLTs), or PEVs left unpulsed or pulsed with SIINFEKL peptide (PP) or OVA for 7 days. (C) Percentage of CD3+CD8+ lymphoproliferative cells after coincubation with DCs, PLTs, or PEVs, unpulsed or pulsed with PP or OVA for 7 days. Data are the mean ± standard error of the mean (SEM); n = 14). **P < .01; ***P < .001; ****P < .0001 vs unpulsed (Friedman test followed by Dunn’s post hoc test for multiple comparisons). NS, nonsignificant. (D) Percentage of CD3+CD8+ lymphoproliferative cells after a 7-day coincubation with PP pulsed DCs or supernatant (surn), depleted of PEVs by ultracentrifugation, left unpulsed or pulsed with PP or OVA. Data are the mean ± SEM (n = 5). **P < .01 vs unpulsed (Mann-Whitney). (E) Proportion of CD3+CD8+ lymphoproliferative cells after a 7-day coincubation with OVA-pulsed PEVs, treated or not with epoxomicin (epoxo) for 2 hours, and PP-pulsed DCs (DC+PP), treated or not with epoxomicin (epoxo). Dashed lines are unstimulated conditions. Data are the mean ± SEM (n = 9 for PEVs and n = 3 for DC). **P < .01 (Wilcoxon). NS, nonsignificant.
As expected, we found that the proportion of lymphoproliferative cells was significantly higher when OT-1 T lymphocytes were incubated with peptide- or native OVA-pulsed DCs or activated platelets (Figure 7B-C). Of particular note, PEVs also supported T-cell proliferation when pulsed with either the SIINFEKL or native OVA (Figure 7B-C). In addition, when PEVs present in the platelet secretome were removed from pulsed conditions by ultracentrifugation, no proliferation was observed, confirming that the pulsed proteins alone, or the platelet secretome devoid of PEVs, cannot support proliferation (Figure 7D). Furthermore, inhibition of the PEV proteasome by epoxomicin before pulsing with native OVA inhibited the ability of PEVs to induce T-cell proliferation (Figure 7E, left). The effect was directed toward the PEV proteasome, as addition of epoxomicin before peptide pulsing at the same concentration used on DCs did not inhibit proliferation (Figure 7E, right). Thus, PEVs are capable of proteosome-dependent processing of native proteins, thereby enabling peptide loading onto MHC-I. PEVs express costimulatory molecules, and their interaction with T lymphocytes promotes lymphocyte cytokine production and proliferation.
Discussion
Megakaryocytes and platelets are emerging as active players in innate and adaptive immunity.7,30,31 The platelet’s role in immunity is mainly confined to the blood circulation, whereas megakaryocytes are localized in bone marrow and lungs. The latter location potentially provides the megakaryocyte with more direct access to airborne pathogens and allergens.32,62,63 In contrast, PEVs can disseminate into organs and tissues, and this dissemination may be due to their small dimensions and the presence of unique surface molecules. In this study, we found that the proteasome and the necessary machinery to process and present antigens to CD8+ T cells are packaged into PEVs by platelets. Thus, PEVs may extend the immune functions played by platelets and megakaryocytes outside the confines of the blood.
PEVs are heterogeneous in terms of surface molecules and their platelet-derived content. The presence of mitochondria within PEVs is well documented,36,37,64 but it was unknown whether other organelles were also transferred from the platelet. The proteasome is much more abundant than mitochondria, at ∼800 000 copies per cell,65 in contrast to ∼3 to 7 mitochondria per platelet.37 Further investigation is necessary to determine whether the presence of multiple organelles within a single vesicle is the result of a specific sorting mechanism or the large size and storage capacity of the vesicles. Nonetheless, we observed that the release of proteasome-containing PEVs requires cytoskeleton remodeling via intact actin microfilament dynamics and that a broad array of platelet agonists induce the release of proteasome-containing PEVs.37
The presence of an extracellular proteasome has already been documented in healthy human blood, and elevated levels have been found in patients who have autoimmune diseases, develop sepsis, or experience trauma.66 Moreover, the 20S proteasome core is present and active within EVs derived from apoptotic endothelial cells and regulates the formation of tertiary lymphoid structure, production of autoantibodies, and graft rejection after transplantation.12 Although some evidence supports that a circulating extracellular proteasome may be transported by EVs, we showed that EVs of platelet origin, among the most abundant EVs in blood, do contain the proteasome. We further suggest, based on our characterization of these EVs, that platelet microvesicles, not exosomes, contain the proteasome. Consistent with this, mass spectrometry analysis of the human PEV proteome identified numerous proteasomal subunits.67-69 These include subunits of the 20S catalytic core and immunoproteasome subunit (PSMB8), subunits of the 11S and 19S regulator, and the 26S proteasome.67-69 Moreover, with calnexin, calreticulin, ERP57, and ERP29, other members of the ubiquitin-proteasome pathway were identified in PEVs, such as members of the E1 and E2 ubiquitin-conjugating enzyme family.67,69 The presence of these proteins and the fact that intact OVA must be ubiquitinated for degradation by the proteasome70 point to the occurrence of functional ubiquitination in PEVs. To our knowledge, there is no evidence of protein TAP-1 and -2 (related to TAP transporter) presence in PEVs. Further investigations are needed to see whether the TAP transporter is present in PEVs and/or whether the processing pathway of the antigen differs in extracellular vesicles, as there is no reported endoplasmic reticulum in PEVs. Thus, although proteomic data point to ubiquitin-proteasome system proteins in PEVs, the present work unequivocally demonstrates its presence and documents that the extracellular proteasome in PEVs is functional and can contribute to antigen processing.
We used complementary approaches and developed an hs-FCM–based assay to detect active proteasome at the single EV level, thereby permitting quantification and assessment of other molecules expressed by the EVs. In particular, the proteasome-containing PEVs also expressed MHC-I and costimulatory molecules, which enabled lymphocyte activation/proliferation and cytokine generation. These findings demonstrate a novel and potentially important role for PEVs in adaptive immunity. Although our work suggests that PEVs are involved in adaptive immunity through antigen presentation, the findings do not necessarily exclude that other cells release proteasome-containing EVs capable of playing this role. Indeed, EVs derived from DCs, B and T lymphocytes, macrophages, and NK cells can perform crosspresentation, suggesting that they also contain the necessary antigen-processing machinery.71-76 Further studies are needed to determine the impact and the importance of PEVs as antigen-presenting elements.
We identified proteasome-containing PEVs in the blood of healthy donors. As most PEVs in blood under healthy conditions are suggested to originate from megakaryocytes,77,78 the latter may also constitutively release proteasome-containing EVs. Moreover, we found that numerous stimuli of human platelets, as well as in vivo stimulation of mouse platelets, induce release of proteasomes in PEVs, suggesting that proteasome release is at least conserved in both humans and mice and takes place via platelet activation. Furthermore, platelets can actively induce immunity against the Plasmodium berghei parasite,26 and megakaryocytes can be infected by dengue virus79 and can also phagocytose Escherichia coli.32 Given their small size, intact microorganisms may not be present inside PEVs, but PEVs may process cytosolic microbial proteins derived from intact platelets/megakaryocytes that lack the ability to enter the lymphatic system. Thus, PEVs may be implicated in immune surveillance and may contribute to presentation of microbial antigens within lymph tissues. Further studies are needed to determine whether exposure to PEVs suffices to establish immunity in vivo, such as to less immunodominant antigens than OVA, or whether costimulation by inflammation or infection is needed to establish sustained immune response.
Self-antigens such as those from mitochondria were identified in a proportion of the proteasome-containing PEVs. Although prior studies showed that mitochondria-containing PEVs are rare in lymph (0.41% ± 0.25% [n = 4] of the PEVs in mouse lymph contain mitochondria)41 in comparison to proteasome-containing PEVs 13.61% ± 4.27% (n = 6), these proportions may be augmented in certain diseases. It would be interesting to determine whether PEVs contribute to the formation of mitochondrial autoantibodies that are described in autoimmune diseases, such as systemic lupus erythematosus.80 Furthermore, the presence of proteasome-containing PEVs in platelet concentrates was not associated with increased risks of ATR or TRALI in a mouse model. It remains to be verified whether the presentation of platelet antigens (eg, CD41 or CD61) by PEVs from PCs contribute to generation of antiplatelet immunity in transfused recipients, although this has been shown with megakaryocytes.56 It also cannot be excluded that PEVs participate in other immune responses, such as autoantibody production or in tissue remodeling, or that they could be used as a platform for cell-based vaccines.81-83 The crosspresentation of PEVs presented herein may allow for new therapeutic possibilities, such as in antitumor or antiviral immunity or inducing cytotoxic immunity by vaccination.84 For example, PEVs have already been proposed as antigen carriers for vaccination,85,86 and our results suggest that these types of PEVs are also endowed with cross-priming properties that offer new prophylactic or therapeutic vaccination.
Human platelets injected into WT mice circulate <2 hours, in contrast to mouse platelets transfused into mice that can circulate for several days.87,88 We thus used mouse PEVs in our transfusion experiments, and yet most were undetectable from the blood circulation after 15 minutes, pointing to their rapid uptake in surrounding tissues. In blood, the main absolute cellular target was the platelets, mostly because platelets outnumber leukocytes, which may suggest that PEVs recycle molecules back to platelets. PEVs were also found in bone marrow, consistent with recent findings that pointed to their role in the stimulation of megakaryocyte biogenesis.43 The main organs that were targeted were the lymphoid organs. Our findings in mouse lymph revealed that proteasome-containing PEVs can circulate in the lymphatic system, potentially explaining their accumulation in lymphoid organs after IV injection. This access to the lymphatic system by proteasome-containing PEVs may reveal a new immune route for PEVs to reach lymphoid organs or infected tissues. Our study highlights the diversity of PEVs and supports the concept that different subtypes of PEVs may play different roles, depending on their cargo and tissue distribution.
Acknowledgments
The authors thank the blood donors and patients who participated in the study; Nicolas Tessandier and Carolanne Gélinas for general technical assistance; and Julie-Christine Lévesque from the Cytometry and Microscopy platform (Centre Hospitalier Universitaire [CHU] de Quebec), and Richard Janvier from the Microscopy platform (Université Laval) for microscopy assistance.
This work was supported by the Natural Sciences and Engineering Research Council (E.B.). E.B. is the recipient of a senior award from the Fonds de Recherche du Québec-Santé (FRQS). This work was also supported by the Établissement Français du Sang (EFS), INSERM, and Université Paris-Est (B.V.) and the Swedish Research Council (Vetenskapsrådet, 2017-01779) (J.W.S.). G.M. received an award from the Canadian Blood Services and from Mitacs and a postdoctoral fellowship from FRQS. A.Z. was the recipient of a postdoctoral fellowship from Swiss National Science Foundation. A.L. has an award from the fonds Pierre Borgeat and from The Arthritis Society (TAS). M.B. is the recipient of a postdoctoral fellowship from TAS.
Authorship
Contribution: E.B., B.V., G.M., and A.Z., conceived and designed the study and wrote the manuscript; G.M., A.L., S.H., M.B., M.M., I.A., M.T., and B.V. performed the experiments, analyzed the data, and participated in manuscript preparation; T.L., J.R., A.K.-R., J.T., H.H.-C., F.C., R.K., J.W.S., M.-J.H., S.G.B., F.P., H.S.O., B.I.F., and M.D. performed experiments, contributed critical biospecimens and analyzed the data; and all authors read and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for G.M. is Division of Hematology and Transfusion Medicine, Lund University, Lund, Sweden.
Correspondence: Eric Boilard, Centre de Recherche du Centre Hospitalier Universitaire de Québec, Faculté de Médecine de l’Université Laval, 2705 Laurier Blvd, Room T1-49, Québec, QC G1V 4G2, Canada; e-mail: eric.boilard@crchudequebec.ulaval.ca; and Benoît Vingert, Établissement Français du Sang, Institut Mondor de Recherche Biomédicale, INSERM U955, 5 rue Gustave Eiffel, 94017 Créteil, France; e-mail: benoit.vingert@efs.sante.fr.
Original data are available by e-mail request to either corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.