Key Points
CAEL-101 is well tolerated in patients with AL amyloidosis.
CAEL-101 leads to rapid and sustained organ responses in patients with AL amyloidosis previously treated with standard therapy.

Abstract
Systemic immunoglobulin light-chain amyloidosis is characterized by pathologic deposition of immunoglobulin light chains as amyloid fibrils in vital organs, leading to organ impairment and eventual death. That the process is reversible was evidenced in an in vivo experimental model in which fibril-reactive chimeric monoclonal antibody (mAb) 11-1F4 directly targeted human light-chain amyloid deposits and affected their removal via a phagocyte-mediated response. To determine the tolerability and potential amyloidolytic effect of this agent (now designated mAb CAEL-101), we conducted a phase 1a/b study involving 27 patients, most of whom had manifestations of organ involvement. This was an open-label study in which phase 1a patients received mAb CAEL-101 as a single intravenous infusion with escalating dose levels from 0.5 mg/m2 to 500 mg/m2 to establish the maximum tolerated dose (MTD). In phase 1b, the antibody was administered as a graded series of 4 weekly infusions. For both phases, there were no drug-related serious adverse events or dose-limiting toxicities among recipients, and the MTD was not reached. The majority of patients had deep hematologic responses but persistent organ disease prior to treatment. Fifteen of 24 patients (63%) who manifested cardiac, renal, hepatic, gastrointestinal, or soft tissue involvement had a therapeutic response to mAb CAEL-101 as evidenced by serum biomarkers or objective imaging modalities with a median time to response of 3 weeks. Infusions of mAb CAEL-101 were well tolerated and, for the majority, resulted in improved organ function, notably for those with cardiac impairment. This trial was registered at www.clinicaltrials.gov as #NCT02245867.
Introduction
Systemic light-chain (AL) amyloidosis is an incurable hematologic malignancy.1,2 Clonal plasma cells in the bone marrow produce soluble κ or λ monoclonal light chains which misfold into insoluble amyloid fibrils. Amyloid fibrils deposit in multiple organs, leading to devastating multi-organ dysfunction. The major determinant of outcomes is the extent of organ involvement.3-5 Cardiac involvement is the most significant negative prognostic factor for overall survival.
The management of AL amyloidosis involves the eradication of monoclonal light-chain-producing clonal plasma cells from the bone marrow. This causes a decrease in the production of light chains, which may permit regression of amyloid deposits and facilitate the restoration of organ function. Only 20% to 25% of patients are eligible to receive high dose melphalan with autologous stem cell transplant (HDM/SCT) because of poor functional status and organ dysfunction.6 However, there have been several advances in antiplasma cell therapies resulting in improved hematologic and organ responses in transplant-ineligible patients over the past few decades. Recently, studies have shown the efficacy of daratumumab, a monoclonal CD38 antibody, in AL amyloidosis.7-10 While significant treatment advances have been made, early mortality rates remain high. In a series of 1551 newly diagnosed AL amyloidosis patients, rates of achieving equal or greater than very good partial response (VGPR) in patients treated with nontransplant regimen improved from 24% to 49% to 56% in 2001-2004, 2005-2009, and 2010-2014, respectively. Organ responses also improved from 21% to 37% to 43% in 2001-2004, 2005-2009, and 2010-2014, respectively. This is largely based on the change from melphalan-based to bortezomib-based regimen. Early mortality of patients not treated with HDM/SCT also decreased from 49% in 2001-2004 to 33% in 2005-2009 and 35% in 2010-2014.11 The still relatively high early mortality is mainly driven by patients with advanced cardiac disease who have sustained and sometimes progressive amyloid deposition in vital organs combined with treatment-related toxicities.12 Currently, no available therapy directly eradicates amyloid deposits which predispose to persistent organ dysfunction. Therapy that leads to more rapid regression of amyloid deposits should improve outcomes in this disease.
The monoclonal antibody (mAb) CAEL-101 is the chimeric form of murine mAb 11-1F4, which binds to a conformational neoepitope contained within the first 18 amino acids of misfolded human immunoglobulin light chains.13-15 It promotes phagocytic destruction and subsequent clearance of amyloid deposits. Consequently, native soluble-free light chains in circulation are spared from destruction. In vivo, both the murine and chimeric form of the mAb expedited dissolution of AL amyloidomas in mice without toxicity.14,15 The specificity of the mAb was confirmed via immunohistochemical and radiographic detection by positron emission tomography-computed tomography (PET-CT) of human AL amyloid deposits in patients who received the mAb intravenously.16 These results led to the development of amyloid fibril-reactive chimeric IgG1 mAb CAEL-101 by the National Cancer Institute’s Biological Resource Branch for a phase 1a/b trial. In this study, we sought to determine the safety, tolerability, and pharmacokinetics (PK) of mAb CAEL-101. We also explored whether organ responses, assessed using validated criteria, could be achieved in AL amyloidosis patients receiving mAb CAEL-101. Here we report the results of the open-label, dose-escalation phase 1a/b study of chimeric IgG1 mAb CAEL-101 (NCT02245867).
Methods
Patients
From September 2014 to April 2017, we enrolled 27 participants (8 in phase 1a and 19 in phase 1b) at Columbia University Medical Center in New York, NY. Five participants in the phase 1a trial also participated in the phase 1b trial. Eligible participants included adults ≥21 years with confirmed histopathologic diagnosis of AL amyloidosis who received prior systemic therapy and had an Eastern Cooperative Oncology Group performance status of ≤3. Patients were excluded for seriously limited cardiac (intraventricular septum thickness >25mm or ejection fraction <40%), renal (creatinine clearance ≤30 mL/min as calculated by the Chronic Kidney Disease Epidemiology Collaboration [CKD-EPI] 2009 formula), or hepatic function (alkaline phosphatase ≥3.0 times the upper limit of normal and total bilirubin ≥3.0 mg/dL), uncontrolled infection, or significant comorbidity. All patients provided written informed consent. Information on prior antiplasma cell therapy, including the number and description of prior therapies and date of last exposure to therapy, were obtained from direct reports of referring providers.
Study design and assessments
This single-arm, open-label, dose-escalation phase 1a/b study was conducted to determine the tolerance, safety, PK, and possible clinical benefit of mAb CAEL-101 in patients with symptomatic AL amyloidosis. The trial protocol included an “up-and-down” dose-escalation design with 2 phases: phase 1a and phase 1b. Patients were allowed to cross over from phase 1a to phase 1b, though the specific patients and dose levels were not prespecified.
In phase 1a, single-patient cohorts received increasing doses of mAb CAEL-101 as a one-time treatment. The starting dose was 0.5 mg/m2, and if tolerated, the doses in subsequent patients were increased in a stepwise fashion to 5, 10, 50, 100, 250, and 500 mg/m2. If a patient experienced dose-limiting toxicity (DLT), defined as any related grade 3 or grade 4 toxicity, then 2 additional patients were sequentially enrolled at the same dose level. All individuals were evaluated on the day of treatment (week 1), weekly for 4 weeks after treatment with mAb CAEL-101 (weeks 2-5), and then 4 weeks later (week 8). Patients who completed phase 1a were permitted to enroll in phase 1b of the study after a washout period of at least 2 months. The patients who crossed over were assessed separately for organ response after each phase (supplemental Table 1, available on the Blood Web site).
In phase 1b, the first patient received 4 weekly infusions of mAb CAEL-101 at 0.5 mg/m2. If tolerated, doses in the subsequent patients were increased to 5, 10, 50, 100, 250, and 500 mg/m2. If a patient experienced a DLT defined as any related grade 3 or 4 toxicity, then 2 additional patients were sequentially enrolled at the same dose level. The maximum tolerated dose (MTD) was defined as the highest dose at which 2 patients had no DLT. When the MTD was reached, an additional 4 patients were enrolled and infused at that dose. The protocol prespecified expansion of the 2 highest dose levels to determine whether there was a dose-dependent response. All individuals were evaluated prior to each mAb CAEL-101 treatment (weeks 1-4), as well as at weeks 5, 8, and 12 (supplemental Table 1). After completion of phases 1a and 1b, patients returned to routine follow-up and monitoring.
The primary objectives were to determine the MTD and evaluate the safety of intravenous mAb CAEL-101. Adverse events were graded according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Safety was assessed through vital signs, clinical assessment of adverse events, and trends in laboratory data.
Secondary objectives included determining PK and assessing organ responses during each study evaluation using the International Society of Amyloidosis’ consensus criteria and the recently clinically validated renal staging and response criteria.17,18 Organ response was only assessed in patients who had organ involvement or measurable disease at screening based on the above-mentioned criteria. Patients were deemed evaluable for cardiac response with N-terminal probrain natriuretic peptide (NT-proBNP) ≥650 pg/mL and for renal response with urine protein ≥500 mg/24 h. Patients with liver involvement were considered evaluable if they had alkaline phosphatase above the upper limit of the institution’s laboratory normal or an abnormal liver size >15 cm by abdominal ultrasound in the absence of decompensated heart failure.19 Cardiac response was defined as a reduction in NT-proBNP of >300 pg/mL and >30% decrease from baseline NT-proBNP.17 In phase 1a, NT-proBNP was measured at baseline and in weeks 1, 2, 3, 4, and 8. In phase 1b, NT-proBNP was measured at baseline and weeks 1, 2, 3, 4, 5, 8, and 12. Kidney response was defined as a ≥30% decrease in 24-hour urine protein to below 500 mg/24 h and no reduction in creatinine clearance by ≥25% below baseline per most recent renal response criteria.17,18 In both phases, the estimated glomerular filtration rate (eGFR) was measured in the same manner as NT-proBNP. However, 24-hour urine measurements were done at baseline and week 8 in phase 1a, with an additional measurement in week 12 in phase 1b. Liver response was defined as a 50% decrease in an abnormal alkaline phosphatase value or a 2 cm decrease in liver size on abdominal ultrasound, whereas a nervous system response was defined by an improvement in electromyogram conduction velocity from an abnormal baseline.19 In addition to standard transthoracic echocardiographic variables such as left ventricular ejection fraction calculated using the Simpson’s biplane method, global longitudinal strain (GLS) was obtained using speckle-tracking echocardiography (TomTec-Arena 1.2, Germany) in phase 1b, as an exploratory substudy. GLS was calculated as an average of 4-, 2-, and 3-chamber based measurements. A more negative change from baseline to the final clinical assessment week indicated improvement. The clinical assessment before the first infusion with mAb CAEL-101 was used as the baseline for the organ response assessment. The best response at any time point after administration of mAb CAEL-101 was also determined. For patients with other organ involvement (gastrointestinal, skin, and soft tissue), objective responses were determined based on improvement in CTCAE grade of symptoms or a measurable reduction in organomegaly on clinical examination. Time to response in weeks was assessed from the week of administration of mAb CAEL-101 to the time of first observed organ response.
PK assessments were performed at screening, on each day of treatment (pretreatment, 1 hour, 2 hours, and 24 hours post start of infusion), and at each weekly assessment for both phase 1a (weeks 2, 3, 4, and 8) and phase 1b (weeks 2, 3, 4, 5, 8, and 12). PK properties were established based on serum concentrations of mAb CAEL-101 using noncompartmental calculation techniques. Measurement of antidrug antibodies against the study agent was not a part of the study.
Study oversight
The study protocol was reviewed and approved by the Institutional Review Board at Columbia University Medical Center and was conducted in accordance with the principles of the Declaration of Helsinki, the International Conference on Harmonization Good Clinical Practice guidelines, and in compliance with the study protocol. All patients provided written informed consent to participate in the study. The study was funded by the National Cancer Institute (NCI Experimental Therapeutics Grant), National Food and Drug Administration (R01 FD005110-01 PI), Columbia Technology Venture, and Caelum Biosciences. The investigators collected the data and can attest to its completeness and accuracy as well as the fidelity of the trial to its approved protocol. The authors of this work analyzed the data, and a review of the analysis was done by Caelum Biosciences.
Statistical analysis
All patients who received at least 1 dose of mAb CAEL-101 were evaluable for toxicity and PK. Descriptive statistics including medians and ranges were used to summarize demographic and baseline patient characteristics. In phase 1b, transthoracic echocardiograms were used to evaluate both left ventricular ejection fraction and GLS at baseline and 12 weeks posttherapy; baseline and posttherapy images were compared in patients with cardiac involvement. Paired student’s t test was also used to compare echocardiographic variables at baseline and at 12 weeks posttherapy.
Results
Patient characteristics
Patients with systemic AL amyloidosis and persistent organ disease were treated in phase 1a (n = 8) and phase 1b (n = 19) with 5 patients enrolled in phase 1a also receiving treatment in phase 1b. Each patient received at least 1 dose of mAb CAEL-101. Baseline demographic and clinical characteristics are shown in Table 1 according to dosing schedule (phase 1a and phase 1b). In the overall population, the median age was 66 years (range, 34-79 years). These patients had similar characteristics to the general AL amyloidosis population with 70.4% (19 of 27) of patients being male and most patients having λ isotype (55.6%, 15 of 27). Median number of organs involved was 2 (range, 1-4). Most patients had cardiac (59.3%, 16 of 27) and renal involvement (48.1%, 13 of 27). The median number of prior antiplasma cell regimen was 2 (range, 1-10). According to the treating physicians, these patients did not require antiplasma cell therapy. The median time since exposure to chemotherapy was 2.6 and 7.4 months (range 0-15.5 months) in phase 1a and 1b, respectively.
Safety
There were no DLTs up to 500 mg/m2 in phase 1a or 1b. The MTD for this study was not reached. No drug-related deaths were observed (supplemental Table 2). The most common treatment-related adverse events in phase 1a included grade 1 to 2 nausea, diarrhea, rash, pruritus, and hyperuricemia in 12.5% (1 of 8 patients). One patient receiving a single dose of 50 mg/m2 developed a grade 2 rash 4 days after mAb CAEL-101 infusion which resolved spontaneously 11 days later. A skin biopsy performed revealed a previously undiagnosed cutaneous amyloidosis with deposits positive for 11-1F4 antibody. In phase 1b, diarrhea, rash, and aspartate transaminase elevation (15.8%; 3 of 19 patients) were the most common adverse events. Of note, 4 weeks after the final dose of mAb CAEL-101 in phase 1b, 1 patient (5.3%) experienced grade 3 pericardial effusion due to advanced cardiac amyloidosis which lasted for 3 months and was clinically observed without intervention. Additionally, the first patient receiving 50 mg/m2 in phase 1b was removed from the study at the investigator’s discretion due to an adverse event thought to be unrelated to mAb CAEL-101. The patient had 1 episode of grade 3 hypotension and vasovagal reaction and was admitted for observation. The patient fully recovered, however, the study team and patient decided to withdraw from the study given the patient’s overall frailty. Another patient was enrolled at the same dose level (50 mg/m2) without incident. The treatment-related adverse events are summarized in Table 2.
PK
Mean serum mAb CAEL-101 concentration-time profiles are shown for phase 1a (Figure 1) and phase 1b (Figure 2). Overall, serum PK of mAb CAEL-101 represents a biphasic disposition due to a rapid distribution phase, slower elimination phase, and long half-life. This is consistent with the PK of other IgG antibodies. The terminal half-life of mAb CAEL-101 in serum after multiple doses was approximately 10 to 16 days. Exposure increased proportionally to dose when the dose was >10 mg/m2 in phase 1a and ≥5 mg/m2 in phase 1b. Peak mAb CAEL-101 levels were obtained after the third dose of 500 mg/m2 in phase 1b but not for other doses.

Mean (SE) PK profiles for mAb CAEL-101 in single ascending dose cohorts by dose level on linear and log-linear scales (phase 1a).
Mean (SE) PK profiles for mAb CAEL-101 in single ascending dose cohorts by dose level on linear and log-linear scales (phase 1a).
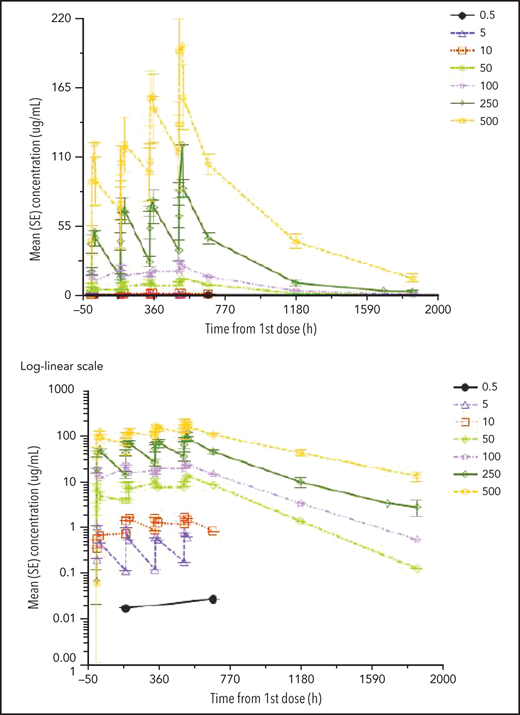
Mean (SE) PK profiles for mAb CAEL-101 in multiple ascending dose cohorts by dose level on linear and log-linear scales (phase 1b).
Mean (SE) PK profiles for mAb CAEL-101 in multiple ascending dose cohorts by dose level on linear and log-linear scales (phase 1b).
Organ responses
Twenty-seven patients were accrued, but 3 patients had no measurable disease. The patient who was removed from the study after 1 dose of mAb CAEL-101 was included in the efficacy analysis. Therefore, 24 patients were evaluable for organ response.
Cardiac response
Of the cardiac-evaluable patients, 67% (8 of 12) showed organ response, 33% (4 of 12) had stable disease, and no patients met criteria for cardiac progression17 (Figure 3). We further evaluated whether cardiac response via NT-proBNP was potentially attributable to drug administration. Following 4 weekly doses of mAb CAEL-101 in phase 1b, there was a statistically significant decrease in NT-proBNP from baseline to week 4 (date of final infusion) of −700 pg/mL (P = .003) (supplemental Figure 1). Additionally, patients with cardiac involvement demonstrated a statistically significant improvement in GLS (−15.58 ± −4.14% pre and −17.37 ± −3.53% post, P = .004) (Figure 4). Subgroup analysis revealed an improvement in GLS in cardiac-evaluable patients with λ subtype amyloidosis (−14.3 ± −4.38% pre and −16.17 ± −3.74% post, P = .02) and a trend toward improvement in patients with κ amyloidosis (−16.60 ± −4.10% pre and −18.16 ± −3.48% post, P = .07).
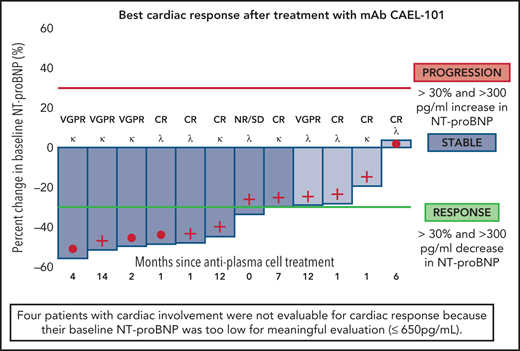
Analysis of percentage change in NT-proBNP from baseline in cardiac-evaluable patients (baseline NT-proBNP of ≥650 pg/mL and at least 1 postbaseline NT-proBNP measurement). Cardiac responders are denoted with deep blue bars (>30% and >300 pg/mL decrease in NT-proBNP). No cardiac progressors (>30% and >300 pg/mL increase in NT-proBNP) were identified. Cardiac stable patients are denoted with light blue bars (neither response nor progression). For each individual patient, time since last exposure to antiplasma cell therapy (months), light chain disease type (κ or λ), and hematologic disease status at the time of study enrollment is reported.
Analysis of percentage change in NT-proBNP from baseline in cardiac-evaluable patients (baseline NT-proBNP of ≥650 pg/mL and at least 1 postbaseline NT-proBNP measurement). Cardiac responders are denoted with deep blue bars (>30% and >300 pg/mL decrease in NT-proBNP). No cardiac progressors (>30% and >300 pg/mL increase in NT-proBNP) were identified. Cardiac stable patients are denoted with light blue bars (neither response nor progression). For each individual patient, time since last exposure to antiplasma cell therapy (months), light chain disease type (κ or λ), and hematologic disease status at the time of study enrollment is reported.

Changes in global longitudinal strain with mAb CAEL-101 in patients with cardiac involvement enrolled in phase 1b (n = 10; blue lines). This figure highlights cardiac response assessment based on measurement of myocardial function at baseline and week 12 using speckle-tracking (TomTec-Arena 1.2) and calculated as an average of 4-, 2-, and 3- chamber based measurements in cardiac response evaluable patients. Nineteen patients were enrolled in the phase 1b study and 10/19 patients were evaluable for cardiac response. NT-proBNP (pg/mL) is included for each patient at baseline (left) and at week 12 (right). An improvement of 2 units (a decrease by −2) is a clinically significant improvement in strain in the AL amyloidosis population. Mean GLS improved significantly in 9/10 patients from −15.58% ± −4.14% at screening to −17.37% ± −3.53% at week 12, P = .004. Pearson correlation coefficient between NT-proBNP response and GLS response (in 8 cardiac evaluable patients) was 0.345, which supports correlation of 2 markers of organ response.
Changes in global longitudinal strain with mAb CAEL-101 in patients with cardiac involvement enrolled in phase 1b (n = 10; blue lines). This figure highlights cardiac response assessment based on measurement of myocardial function at baseline and week 12 using speckle-tracking (TomTec-Arena 1.2) and calculated as an average of 4-, 2-, and 3- chamber based measurements in cardiac response evaluable patients. Nineteen patients were enrolled in the phase 1b study and 10/19 patients were evaluable for cardiac response. NT-proBNP (pg/mL) is included for each patient at baseline (left) and at week 12 (right). An improvement of 2 units (a decrease by −2) is a clinically significant improvement in strain in the AL amyloidosis population. Mean GLS improved significantly in 9/10 patients from −15.58% ± −4.14% at screening to −17.37% ± −3.53% at week 12, P = .004. Pearson correlation coefficient between NT-proBNP response and GLS response (in 8 cardiac evaluable patients) was 0.345, which supports correlation of 2 markers of organ response.
Renal response
In a best response analysis for renal-evaluable patients, 20% (2 of 10) showed organ response. Sixty percent (6 of 10) had stable disease. This group also had improved eGFR. The remaining 20% of patients (2 of 10) met criteria for progression of renal disease based on reduction in eGFR despite having improvements in proteinuria >30% from baseline (Figure 5).

Analysis of best response of percentage change in 24-hour urine protein from baseline in renal-evaluable patients (baseline 24-hour urine protein of >500 mg/24 h and at least 1 postbaseline 24-hour urine protein measurement). Renal responders are denoted with deep blue bars (≥30% decrease in proteinuria or fall in 24-hour urine protein below 500 mg/24 h). Renal stable patients are denoted with light blue bars (neither response nor progression). Renal progressors are denoted by red bars (≥25% decrease in eGFR). For each individual patient, time since last exposure to antiplasma cell therapy (months), light chain disease type (κ or λ), and hematologic disease status at the time of study enrollment is reported.
Analysis of best response of percentage change in 24-hour urine protein from baseline in renal-evaluable patients (baseline 24-hour urine protein of >500 mg/24 h and at least 1 postbaseline 24-hour urine protein measurement). Renal responders are denoted with deep blue bars (≥30% decrease in proteinuria or fall in 24-hour urine protein below 500 mg/24 h). Renal stable patients are denoted with light blue bars (neither response nor progression). Renal progressors are denoted by red bars (≥25% decrease in eGFR). For each individual patient, time since last exposure to antiplasma cell therapy (months), light chain disease type (κ or λ), and hematologic disease status at the time of study enrollment is reported.
Other clinical improvements
Three patients with involvement of other organs had clinical improvement while on treatment. One patient with gastrointestinal involvement treated with 5 mg/m2 in phase 1a demonstrated improvement in diarrhea, nausea, and postprandial abdominal pain (CTCAE grade 3 to 1). Another patient with debilitating hand arthritis from soft tissue involvement had subjective improvement in the size of soft tissue lesions and range of motion of the hands following treatment with 500 mg/m2 (CTCAE grade 3 to 1). Both patients with κ amyloidosis and liver involvement experienced a decrease in liver size (as revealed by ultrasound in 1 case and by clinical examination in the other), both after receiving 500 mg/m2. The first patient had cardiac amyloidosis not evaluable for response due to NT-proBNP <650 pg/mL but experienced a 1.6 cm reduction in liver size (16 cm pretreatment to 14.4 cm posttreatment) after 1 infusion of mAb CAEL-101 in phase 1a. The second patient had no evidence of cardiac involvement and experienced a 2.1 cm reduction in liver size (19.4 cm pretreatment to 17.3 cm posttreatment) at week 8 of treatment in phase 1b.
Overall organ responses
In both phase 1a and 1b, organ responses were noted in evaluable patients receiving >5 mg/m2 of the antibody. There were no organ responses at lower doses. The overall organ response rate was 63% (15 of 24 evaluable patients) in those receiving at least 1 dose of mAb CAEL-101 independent of the free light chain subtype or hematologic status at enrollment.
Time to response
The median time to response was 3 weeks after the first infusion of mAb CAEL-101. Organ response was observed regardless of prior hematologic response to antiplasma cell therapy, including 1 patient with cardiac λ amyloidosis who had 6 prior treatments.
Discussion
Achieving meaningful organ responses in patients with light-chain amyloidosis is essential for symptom reduction and improving overall survival. Consequently, there is a critical need for therapies that target and rapidly dissolve amyloid deposits.
The findings of this phase 1a/b study confirm that treatment with mAb CAEL-101 is well tolerated with the potential to improve biomarkers associated with organ dysfunction in patients with AL amyloidosis. There was no DLT observed up to 500 mg/m2. The toxicities associated with mAb CAEL-101 were generally mild. The most notable skin toxicities occurred in patients with previously unknown cutaneous amyloidosis, likely due to direct binding of mAb CAEL-101 to skin amyloid deposits. There was subsequent clearance within 1 to 2 weeks of starting infusions. Adverse reactions were easily managed with observation or supportive care. The response rate was 67% and 20% in cardiac and renal-evaluable patients, respectively. These organ response rates are improved in comparison with those reported in published data sets.20,21 Because cardiac involvement drives poor outcomes in AL amyloidosis, a growing body of literature suggests that objective assessment of cardiac response to new therapies is imperative. Several studies have suggested that GLS independently predicts survival in AL amyloidosis and offers objective clinical information earlier than standard serological markers and transthoracic echocardiogram.22-24 In this context, our exploratory analysis included serial GLS measurements via speckle tracking echocardiography. This is the first clinical trial to demonstrate significant improvement in cardiac function via GLS after treatment with amyloid fibril-targeted therapy. Moreover, organ responses occurred early, within 3 weeks after the first infusion of mAb CAEL-101. This is contrary to the median time to organ response of 10.4 months after standard therapy.20 If confirmed in future studies, this demonstrates a true reduction in toxic intermediates and pathogenic amyloid deposits. The subgroup analysis of phase 1b patients showing deeper organ responses during infusion weeks suggests that there may be reaccumulation of amyloid deposits when the drug is stopped (supplemental Figure 1). This is possibly due to free light chains which remain in circulation in patients without adequate hematologic response to therapy. These mechanisms likely overcome the previous on-target effects of the drug as it is cleared from the body. Improvement in organ function during therapy with mAb CAEL-101 followed by a subsequent decline in organ function after treatment could also suggest that additional infusions are needed to induce continuous clearance of amyloid deposits from involved organs.
Because antiplasma cell therapy may require months to induce an organ response, it is possible that responses attributed to CAEL-101 represent the effects of prior lines of therapy. However, none of the responders had improved biomarkers prior to the administration of CAEL-101 despite multiple screening and baseline measurements. It is then very unlikely that all the responders would have a putative delayed response to antiplasma cell therapy immediately after CAEL-101 was administered. While imaging parameters such as a decrease in liver size on ultrasound and global longitudinal strain on echocardiogram showed clinically significant improvement, our study was not blinded (due to the nature of its design), and so operators were cognizant of treatment being administered. Furthermore, we were unable to control for factors that potentially affect biomarker levels. For example, troponin and NT-proBNP levels can be altered by sodium balance, fluid shifts, diuretic use, and renal impairment. These confounders were not assessed separately in this study. Nevertheless, these biomarkers have the most robust data for predicting changes in organ dysfunction and overall survival.4,17 Randomized phase 2 and 3 trials should provide better means of controlling for confounders. Hematologic response is an essential endpoint for AL amyloidosis trials because it is an indirect determinant of persistent generation of toxic free light chains and new amyloid deposits. However, hematologic response was not measured in this trial. CAEL-101 and antiplasma cell therapy work by different mechanisms, so traditional organ response criteria may need to be reconsidered. Specifically, an incomplete or lack of response to chemotherapy may not preclude a meaningful organ response to anti-amyloid-directed therapy.
The simultaneous induction of hematologic and organ response using therapies with different mechanisms of action may allow for HDM/SCT in those who were previously ineligible and improve survival of this invariably fatal disease. This concept has spurred the development of other anti-amyloid fibril therapies. First, a humanized antibody to serum amyloid P component (SAP, a glycoprotein found on all amyloid deposits) was shown to reduce visceral secondary amyloid deposits in mouse models. In a phase 1 clinical trial, the combination of SAP antibody (anti-SAP) and a compound depleting circulating SAP (R-1-{6-[R-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl} pyrrolidine-2-carboxylic acid, CPHPC) led to a reduction in renal and hepatic amyloid load.25 A phase 2 trial (NCT03044353) examining the efficacy of this combined therapy was terminated due to an unfavorable risk-to-benefit ratio. Another humanized mAb, NEOD001, was originally developed against AA amyloid but was found to have high affinity for AL amyloid fibrils in vitro. In vivo, NEOD001 induced phagocyte-mediated clearance of AL amyloid in mouse models.26,27 Given these preclinical findings, it was hypothesized that NEOD001 directly targets human AL amyloid and promotes clearance of amyloid fibrils. NEOD001 was well tolerated and elicited organ responses within 4 to 24 months in the phase 1 trial.28 However, the phase 2b PRONTO study did not meet its endpoints as there was no significant difference between NEOD001 and control arms in the composite endpoint of cardiac hospitalizations and all-cause mortality. The phase 3 VITAL study of NEOD001 was prematurely stopped based on a futility analysis, although a posthoc analysis suggests a potential survival benefit in patients with Mayo Stage IV cardiac amyloidosis.29
Unlike NEOD001, the specificity of mAb CAEL-101 was confirmed in human subjects by different methods prior to this phase 1a/b clinical trial. In studies examining the feasibility of amyloid imaging, PET-CT in human subjects revealed uptake of the 124I-labeled mAb in areas deemed to contain AL amyloid deposits. These areas were biopsied and subjected to Congo red and immunohistochemical staining with the mAb.16 The identical patterns of staining with Congo red and mAb CAEL-101 proves direct binding and detection of AL amyloid by mAb CAEL-101.
Overall, the present study demonstrates that mAb CAEL-101 has an acceptable safety profile and is likely to be clinically active in patients with AL amyloidosis. These data provide a platform for future studies to define the clinical activity of mAb CAEL-101 at doses >500 mg/m2 and in combination with antiplasma cell therapy. Multicenter phase 2 and 3 trials are underway.
Acknowledgments
This work was funded by the National Cancer Institute Experimental Therapeutics (NExT) grant, FDA R01 FD005110-01 PI, and Columbia Technology Ventures at Columbia University.
Authorship
Contribution: A.S. designed the study, edited the manuscript, and approved the final version; S. Lentzsch designed the study, performed the research, enrolled, subjects, edited and approved the final version of the manuscript; C.V.E. collected data, analyzed the data, performed statistical analysis, and wrote the manuscript; A.E. analyzed the data, edited the manuscript, and approved the final version; N.R., D.B., M.S.M., J.R., S.S., M.M., and S. Leng performed research, edited the manuscript and approved the final version of the manuscript; and A.S. and S. Leng approved the final version of the manuscript.
Conflict-of-interest disclosure: A.S. is a consultant for Caelum Biosciences and a recipient of stock options. S. Lentzsch has received compensation as a member of the scientific advisory boards of Janssen, Celularity, AbbVie, GSK, Takeda, Karyopharm, Sanofi, and Oncopeptides, and received research funding from Karyopharm and Sanofi. She was the chief scientific advisor, board of directors' member and a shareholder of Caelum Biosciences. M.M. is a consultant for Ossium Health Inc and a recipient of sponsored research agreements. In addition, M.M. is the spouse of S. Lentzch and both have their conflicts of interest by proxy.
The current affiliation for C.V.E. is Boston Medical Center, Section of Hematology and Oncology, Department of Internal Medicine, Boston University, Boston, MA.
Correspondence: Andrew Eisenberger, Herbert Irving Pavilion, 161 Fort Washington Ave, Floor: Garden Level, New York, NY 10032; e-mail: abe6@cumc.columbia.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.