Key Points
A fusion protein with an anticoagulant peptide linked to annexin V binds extracellular vesicles in circulation
This fusion protein prevented consumptive coagulopathy and improved outcomes of mice subjected to severe traumatic brain injury.

Abstract
Severe traumatic brain injury (TBI) often causes an acute systemic hypercoagulable state that rapidly develops into consumptive coagulopathy. We have recently demonstrated that TBI-induced coagulopathy (TBI-IC) is initiated and disseminated by brain-derived extracellular vesicles (BDEVs) and propagated by extracellular vesicles (EVs) from endothelial cells and platelets. Here, we present results from a study designed to test the hypothesis that anticoagulation targeting anionic phospholipid-expressing EVs prevents TBI-IC and improves the outcomes of mice subjected to severe TBI. We evaluated the effects of a fusion protein (ANV-6L15) for improving the outcomes of TBI in mouse models combined with in vitro experiments. ANV-6L15 combines the phosphatidylserine (PS)-binding annexin V (ANV) with a peptide anticoagulant modified to preferentially target extrinsic coagulation. We found that ANV-6L15 reduced intracranial hematoma by 70.2%, improved neurological function, and reduced death by 56.8% in mice subjected to fluid percussion injury at 1.9 atm. It protected the TBI mice by preventing vascular leakage, tissue edema, and the TBI-induced hypercoagulable state. We further showed that the extrinsic tenase complex was formed on the surfaces of circulating EVs, with the highest level found on BDEVs. The phospholipidomic analysis detected the highest levels of PS on BDEVs, as compared with EVs from endothelial cells and platelets (79.1, 15.2, and 3.5 nM/mg of protein, respectively). These findings demonstrate that TBI-IC results from a trauma-induced hypercoagulable state and may be treated by anticoagulation targeting on the anionic phospholipid-expressing membrane of EVs from the brain and other cells.
Introduction
Trauma is a leading cause of preventable death. More than 50% of trauma deaths are caused by uncontrolled hemorrhage due to primary vascular injury and secondary coagulopathy.1,2 Trauma-induced coagulopathy is commonly caused by significant blood loss (hemorrhagic shock), hemodilution due to fluid resuscitation, hypothermia and metabolic acidosis caused by microvascular hypoperfusion and tissue ischemia, dysfunctional platelets and coagulation, and hyper-fibrinolysis.3-7 Traumatic brain injury (TBI) has the highest mortality among trauma patients, with many dying of secondary injuries, including dysfunctional hemostasis and the resultant coagulopathy. TBI-induced coagulopathy (TBI-IC) is common,8-10 despite the fact that patients with isolated TBI lack key factors that cause coagulopathy in patients with trauma to the body and limbs and hemorrhagic shock.
TBI-IC develops systemically but is often manifested as secondary or delayed intracranial and intracerebral hematoma. TBI-IC is commonly diagnosed by laboratory findings in peripheral blood samples of patients (eg, INR and thromboelastogram)11, suggesting that it disseminates systemically. This systemic response to a localized cerebral injury strongly suggests that injured brains release causal factors into the systemic circulation. Our studies have identified brain-derived extracellular vesicles (BDEVs) as key initiating and disseminating factors that induce a systemic hypercoagulable state that rapidly turns into consumptive coagulopathy.12,13 These BDEVs express abundant and highly procoagulant anionic phospholipids such as phosphatidylserine (PS) and tissue factor (TF) that cause a rapid hypercoagulable state when they are infused into noninjured mice.12,14 BDEVs, including extracellular mitochondria, also activate endothelial cells,12,15 resulting in the endotheliopathy that has been widely attributed to secondary injuries induced by TBI and trauma to the body and limbs.16 These observations led us to hypothesize that circulating extracellular vesicles (EVs) serve as mini platforms on which coagulation is initiated and propagated during acute TBI. In support of this hypothesis, we have shown that the apoptotic scavenge factor lactadherin17 improves outcomes of TBI mice by removing these procoagulant EVs through phagocytosis.13 More importantly, we observed that the PS-binding C1C2 domain of lactadherin was less efficacious in reducing the TBI-induced hypercoagulable state, suggesting that blocking PS alone is insufficient to prevent TBI-IC. It is this observation that led us to design the current study to test the fusion protein ANV-6L15, which links a Kunitz protease inhibitor module (6L15) to annexin V (ANV) for blocking TBI-induced consumptive coagulopathy caused by PS-expressing EVs and injured cells in mouse models.
Materials and methods
ANV-6L15 is a recombinant fusion protein that fuses a Kunitz protease inhibitor module, 6L15, into ANV.18 The ANV enables the fusion molecule to bind anionic phospholipids, such as PS exposed on the surfaces of apoptotic cells and EVs, to deliver the anticoagulant activity of 6L15.19 6L15 is an aprotinin variant that preferentially inhibits TF-FVIIa complex (Ki ∼0.2 nM) to suppress the initiation and propagation of the extrinsic coagulation.20 ANV cDNA was first amplified from human placental mRNA with 2 modifications: removing the stop code and conversing Cys316 to Ala. The synthetic genes encoding 6L15 were constructed by the annealing and ligation of 3 pairs of overlapping oligonucleotides.20 The fusion protein contains 378 amino acids, 319 from ANV and 59 from 6L15 (Met1-Ala59), and it has a molecule mass of 42 kDa. ANV-6L15 was expressed in Escherichia coli DH5α and purified using Q-Sepharose chromatography followed by absorption to PS-containing liposomes. Purified ANV-6L15 is about 6000-fold more potent than 6L15 in the inhibition of TF-induced coagulation in vitro.18 To ensure that there was no significant difference in osmolality between the ANV-6L15 solution (treatment) and the vesicle buffer (control), we measured their viscosities using a viscometer (Thermo HAAKE). The viscosities of ANV-6L15 at 2 µg/mL (therapeutic dose) and 20 µg/mL were 0.716 ± 0.012 centipoise (cp) and 0.708 ± 0.008 cp, respectively, indistinguishable from the viscosity of the vehicle buffer (0.721 ± 0.008 cp).
Mouse model of fluid percussion injury
The study was conducted on adult male C57BL/6J mice (12 to 16 weeks old and 22 to 25 g; The Jackson Laboratory, Bar Harbor, ME),12 as described in the supplemental Methods (available on the Blood Web site). This protocol was approved by the Institutional Animal Care and Use Committee of the Bloodworks Research Institute. Mice were randomly chosen to receive fluid percussion injury (FPI) at 1.9 ± 0.2 atm or to undergo identical surgery without FPI (sham). The FPI mice were randomly assigned to receive either 90 µg/kg of ANV-6L15 (∼2 µg/mL) or an equal volume of the vehicle phosphate-buffered saline (PBS) through the tail vein 30 minutes after FPI. This dosage was chosen based on a dose-titration experiment for blocking 50% of BDEV-induced coagulation (supplemental Figure). For comparison with ANV-6L15, a subgroup of FPI mice received 90 µg/kg of ANV (ThermoFisher Scientific, Waltham, MA).
The mice were studied for the following outcomes: (1) EVs in blood samples collected through retro-orbital veins (100 µL per draw), measured using flow cytometry; (2) 3-day survival and neurological function, measured using the modified neurological severity score (mNSS)12,21; (3) vascular permeability, measured using the Evans blue extravasation test12; (4) tissue edema, determined by the water content of the brain and lungs (supplemental Methods); (5) perivascular bleeding, tissue edema, and intravascular fibrin deposition, detected by the histopathology of the brain and lungs (supplemental Methods); (6) magnetic resonance imaging (MRI) scanned at 3 hours after TBI; (7) the hypercoagulable state, measured by thrombin generation, clotting time, plasma fibrinogen, and D-dimer (supplemental Methods); and (8) hemostasis, measured by tail bleeding (supplemental Methods).22,23 To minimize the confounding influence of blood draws and other technical manipulations on the outcome assessments of the mice, mNSS, MRI, histopathology, tail bleeding, and vascular permeability evaluations were made on separate groups of mice and analyzed as independent variables.
We also extracted mitochondria from the brains of mice expressing mitochondria-specific green fluorescent protein (GFP) (C57BL/6J-Tg[CAG-Cox8/EGFP]49Rin, RIKEN, Tokyo, Japan)24 using a commercial kit according to the manufacturer’s instructions (ThermoFisher Scientific). Morphologically intact and metabolically active extracellular mitochondria (exMTs) were verified by detecting intrinsic GFP and labeling with a Cy5-MetoTracker (ThermoFisher Scientific), as we previously reported.21 GFP+ exMTs (1 × 105/µL × 100 µL per mouse) were infused into the noninjured C57BL/6J mice through the tail vein with either ANV-6L15 or an equal volume of the vehicle PBS. Blood samples were collected 30 minutes after injection to detect GFP+ exMTs on platelets marked by an APC-anti-CD41a antibody (1 µg/mL; ThermoFisher Scientific); their activation was detected by an APC anti-CD62p antibody (2 µg/mL, BD Biosciences). This experiment was designed to: (1) validate the binding of BDEVs to platelets because the anionic phospholipid cardiolipin exposed on the surface of exMTs binds platelets to the lipid receptor CD36; and (2) explore the role of ANV-6L15 in blocking the exMT-induced activation of platelets, which contributes significantly to TBI-IC.21,25
Coagulation
We used 3 complementary assays to measure the state of coagulation and fibrinolysis. First, thrombin generation was measured using a FluCa Kit on Thrombinoscope BV (Diagnostica Stago, Asnières-sur-Seine, France; supplemental Methods).12 Second, clotting time was measured using an STA-Procoag-PPL Kit to test the ability of EVs to clot phospholipid-depleted plasma in the presence of 0.02 U/mL of FXa.13,22 Third, an enzyme-linked immunosorbent assay was used in accordance with the manufacturer’s instructions to measure the plasma levels of fibrinogen (Abcam, Cambridge, United Kingdom) and D-dimer (LifeSpan Biosciences, Seattle, WA).12
Flow cytometry
We used flow cytometry (LSR II; Beckon Dickinson, San Jose, CA) to detect EVs in the peripheral blood of TBI and sham mice, as previously described.12,13,22 In brief, 10 µL of blood sample in 0.32% sodium citrate (the final concentration) was diluted with PBS and incubated with antibodies for 20 minutes at room temperature (RT). The samples were then fixed with 5% paraformaldehyde in PBS and analyzed using flow cytometry. EVs were first identified by their size (<1 μm), measured using standard microbeads of 0.5, 0.9, and 3μm (Biocytex, Marseille, France), and then by their expression of specific cell markers in the gate setting illustrated in supplemental Figure 2. The antibodies used to detect cell markers included: (1) an FITC-conjugated glial fibrillary acidic protein (GFAP) antibody (1 µg/mL; eBioscience, San Diego, CA) for detecting glial cell EVs12; (2) an eFluor 450-conjugated rat anti-mouse CD41a antibody (1 µg/mL; Invitrogen, Carlsbad, CA) for detecting platelet EVs (pEVs); (3) an eFluor 450-conjugated rat anti-mouse CD144 antibody (4 µg/mL; Invitrogen) for detecting endothelial EVs (eEVs); (4) a PE-conjugated monoclonal CD45 antibody (2 µg/mL; Invitrogen) for detecting leukocyte EVs (leuEVs); (5) goat anti-mouse FVII (0.4 µg/mL; Bio-Techne, Minneapolis, MN), rabbit anti-mouse TF (0. 8 µg/mL; Bioss, Woburn, MA), rabbit anti-mouse FXa (5.0 µg/mL; Abcam), and Alexa Fluor 647-conjugated anti-fibrinogen (15 µg/mL; ThermoFisher Scientific) antibodies for detecting the EV-bound coagulation factor VII, TF, activated factor Xa, and fibrinogen; (6) PE-conjugated ANV (5 µL per test; BD Biosciences) for binding PS exposed on the surface of EVs in the presence of calcium along with antibodies against cell markers; (7) an FITC-conjugated anti-ANV antibody (5 µL per sample, 1/100 dilution; Invitrogen) for detecting EV-bound ANV-6L15; (8) an FITC-conjugated rat anti-mouse CD62p antibody (2 µg/mL; BD Biosciences, San Jose, CA) for detecting platelet activation. Appropriate isotype IgGs were used as negative controls. All buffers were filtered with a 0.1-μm filter (EDM Millipore, Billerica, MA) before use to reduce small particle contamination when detecting EVs.
Vascular permeability
MRI
Mice subjected to FPI or sham surgery were scanned for hematoma 3 hours after injury on a 1.0 T Bruker ICON MRI system (Bruker BioSpin, Billerica, MA). Scout images were obtained first to localize the region of interest, which was centered on the FPI impact site. Axial T2-weighted images were then acquired using rapid acquisition with a relaxation enhancement (RARE) sequence (ratio of the volume 20 × 20 mm2, matrix = 140 × 140, slice = 0.8 mm, slice number = 15 TR = 3563.5 ms, TE = 110.0 ms, RARE factor = 8, excitation angle = 90°, refocusing angle = 180°, scan time = 10 minutes). Hematoma volume was measured using the signal intensity of the axial T2-weighted images. The hematoma boundary was traced manually using a RadiAnt Dicom Viewer (Medixant, Poznan, Poland), and the hematoma area of each section was multiplied by the section thickness to yield the hematoma volume.26
Immunoblot
EVs concentrated from the plasma samples of TBI mice were washed with PBS, fixed in 5% paraformaldehyde, solubilized in 1% SDS sample buffer, and subjected to polyacrylamide gel electrophoresis. After transfer to a polyvinylidene fluoride membrane, EV-bound ANV-6L15 was detected by an HRP-conjugated anti-ANV antibody (1/1000 dilution; St John's Laboratory, London, United Kingdom). In a separate experiment, BDEVs made from the brains of noninjured mice (supplemental Methods12) were incubated with ANV-6L15 (0.3 µg/µL, final concentration) for 15 minutes at RT. EV-bound ANV-6L15 was detected by the anti-ANV antibody.
Mass spectrometry analysis
We used mass spectrometry to quantify the phospholipid PS and phosphatidylcholine (PC) content on EVs from TBI and sham mice. EVs were concentrated from plasma pooled from 5 mice in each experimental group.12,13 Phospholipids were extracted using liquid chromatography-mass spectrometry (LC-MS) grade 2-propanol (final concentration 95% V/V; Fisher Scientific) and diluted 1:1 (v/v) with 10 mM of LC-MS-grade ammonium acetate in methanol/water (90:10, v/v; Sigma Aldrich, St Louis, MO). They were then mixed with an internal standard mixture (Odd-Chained LIPIDOMIX and SM 16:0-d31-18:1; Avanti Polar Lipids, Alabaster, AL). The total proteins in each sample were measured using the Bradford protein assay (Bio-Rad, Hercules, CA) before lipid extraction using bovine serum albumin (BSA) as standard.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed using the mass spectrometer QTrap 6500 (AB Sciex, Framingham, MA) coupled with an ultraperformance liquid chromatographer (UPLC; Acquity I-Class; Waters). The extracted phospholipids were separated by LC using a C8 column (Acquity BEH C8, 1.7 µm, 2.1 × 100 mm; Waters) and detected in multiple reaction monitoring mode (MRM). Specific phospholipids were quantified by peak area relative to the internal standard using Analyst version 1.6.2 and MultiQuant version 2.1.1 (AB Sciex) as nM/mg of proteins.
Statistical analysis
The data were presented as mean ± standard error of the mean (SEM) for continuous variables and as percentages for categorical variables, and they were analyzed using SigmaPlot (v11.2, SYSTAT Software, San Jose, CA). The quantitative data were first analyzed using the Shapiro-Wilk normality test for data distribution. A Student t test was used to compare the 2 groups of quantitative variables, and 1-way analysis of variance (ANOVA) or Kruskal-Wallis ANOVA on ranks test was used for Bonferroni posthoc comparisons. The survival index was analyzed using the Kaplan-Meier plot.
Further technical details can be found in the supplemental Methods for (1) the mouse model of fluid percussion injury, (2) the measurement of vascular permeability in vivo, (3) thrombin generation assay, (4) tail bleeding, and (5) production of BDEVs from noninjured brains subjected to freeze-thaw injury.
Results
ANV-6L15 improved outcomes for mice subjected to severe TBI
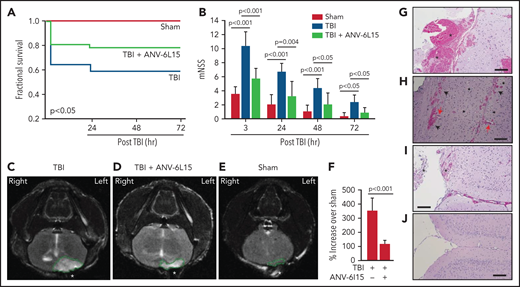
Mice exposed to FPI at 1.9 ± 0.2 atm had an overall mortality of 41.8%, and all deaths occurred between 4 and 24 hours after injury (Figure 1A). The surviving TBI mice developed neurological deficits that were most severe in the first 6 hours and improved progressively thereafter (Figure 1B). ANV-6L15 given as a single bolus dose of 90 µg/kg IV reduced mortality by 56.8% and significantly improved the neurological function of the TBI mice (Figure 1A,B). The body temperature was comparable among the sham mice and the TBI mice receiving ANV-6L15 and those receiving PBS, but the TBI mice became hypertensive and developed bradycardia at 3 hours postinjury, which was significantly improved in those receiving ANV-6L15 (Table 1). The TBI mice also had reduced pO2 and increased pCO2, indicating systemic hypoxia, which was reversed in the TBI mice receiving ANV-6L15 (Table 1). The sham mice also had mildly reduced pO2 without increasing pCO2, likely caused by repeated anesthesia (first for craniotomy and second for FPI), as previously reported.27 Consistent with the hemorrhagic phenotype, TBI mice had lower red blood cell (RBC) counts, hematocrits, and hemoglobin, which were improved by ANV-6L15 (supplemental Table 2).
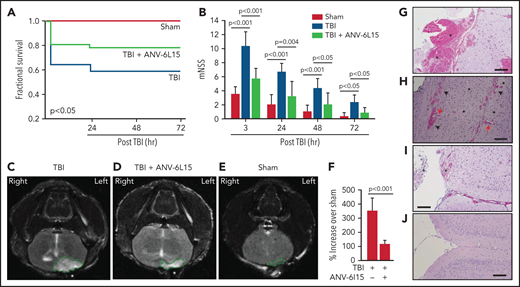
ANV-6L15 increased survival, improved neurological function, and reduced hematoma in TBI mice. C57BL/6 male mice were subjected to FPI and received 90 µg/kg of ANV-6L15 by IV 30 minutes postinjury or underwent sham surgery. (A-B) Their 3-day survival was measured by Kaplan Meier survival analysis (A) (n = 12 mice per group), and their neurological functions were evaluated using mNSS (B) (n = 12 mice per group, 1-way ANOVA). (C-D) TBI and sham mice also received MRI scans 3 hours after injury. FPI induced large subdural hematoma (the white area enclosed by the green circle; asterisk indicates the site of FPI) and intraventricular hemorrhage (C), which was significantly reduced in TBI mice receiving ANV-6L15 (D). (E-F) Small cerebral lesions were also detected in sham mice (E), and their volume was set as the baseline on which the hematoma volume of TBI mice was calculated (F) (n = 6 mice per group, paired Student t test). (G-H) Representative images of H&E stained cerebral tissue from TBI mice show a large subdural hematoma (asterisk; scale bar, 50 µm) (G), substantial intracerebral (black arrows) and perivascular hemorrhage (red arrows) (H), and multiple foci of cerebral edema (black star; scale bar, 50 µm). (I-J) These changes were less severe in TBI mice receiving ANV-6L15 (I) (asterisk denotes subdural hematoma, scale bar, 50 µm), and were not observed in sham mice (J).
ANV-6L15 increased survival, improved neurological function, and reduced hematoma in TBI mice. C57BL/6 male mice were subjected to FPI and received 90 µg/kg of ANV-6L15 by IV 30 minutes postinjury or underwent sham surgery. (A-B) Their 3-day survival was measured by Kaplan Meier survival analysis (A) (n = 12 mice per group), and their neurological functions were evaluated using mNSS (B) (n = 12 mice per group, 1-way ANOVA). (C-D) TBI and sham mice also received MRI scans 3 hours after injury. FPI induced large subdural hematoma (the white area enclosed by the green circle; asterisk indicates the site of FPI) and intraventricular hemorrhage (C), which was significantly reduced in TBI mice receiving ANV-6L15 (D). (E-F) Small cerebral lesions were also detected in sham mice (E), and their volume was set as the baseline on which the hematoma volume of TBI mice was calculated (F) (n = 6 mice per group, paired Student t test). (G-H) Representative images of H&E stained cerebral tissue from TBI mice show a large subdural hematoma (asterisk; scale bar, 50 µm) (G), substantial intracerebral (black arrows) and perivascular hemorrhage (red arrows) (H), and multiple foci of cerebral edema (black star; scale bar, 50 µm). (I-J) These changes were less severe in TBI mice receiving ANV-6L15 (I) (asterisk denotes subdural hematoma, scale bar, 50 µm), and were not observed in sham mice (J).
The TBI mice developed a large subdural hematoma and intraventricular bleeding, but the hematoma volume was significantly reduced in those receiving ANV-6L15 (Figure 1C-D,F). The sham mice also developed minor cerebral injuries but not hematoma (Figure 1E), likely caused by the surgical procedures. Hematoxylin and eosin (H&E) stains of brain sections detected not only hematoma (Figure 1G) but also intracerebral hemorrhage with multifocal tissue edema (Figure 1H) in the TBI mice. The bleeding was much less severe in the TBI mice receiving ANV-6L15 (Figure 1I). Together, these results suggest that the TBI mice developed acute conditions that mimicked the clinical presentation of patients with severe close TBI, and ANV-6L15 reduced the severity of these conditions by preventing the accumulation of intracranial hematoma.
ANV-6L15 reduced vascular permeability and cerebral edema in TBI mice
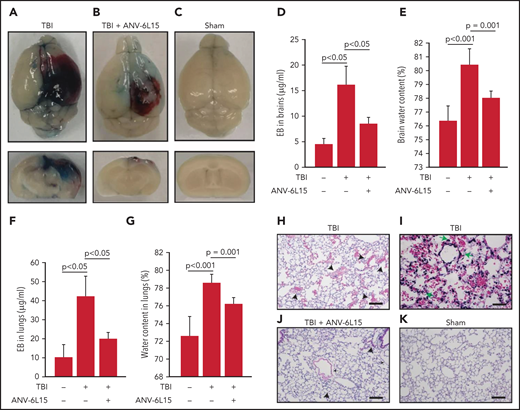
TBI mice receiving ANV-6L15 also developed less endothelial permeability in the brain, as measured by Evans blue extravasation (Figure 2A-D), and reduced tissue edema, as measured by cerebral water content (Figure 2E). The vascular permeability and resultant interstitial tissue edema were also detected in the lungs of TBI mice and reduced in ANV-6L15-treated TBI mice (Figure 2F-G). H&E stains of lung tissue detected diffused alveolar bleeding and microvascular thrombi in TBI mice, which were largely absent from the TBI mice receiving ANV-6L15 (Figure 2H-K). Neural tissue emboli were sporadically detected within the pulmonary vasculature of TBI mice (supplemental Figure 3), consistent with findings from TBI patients.28,29 Together, these results indicate that ANV-6L15 reduced injury-induced vascular permeability not only in the brain but also in the lungs.
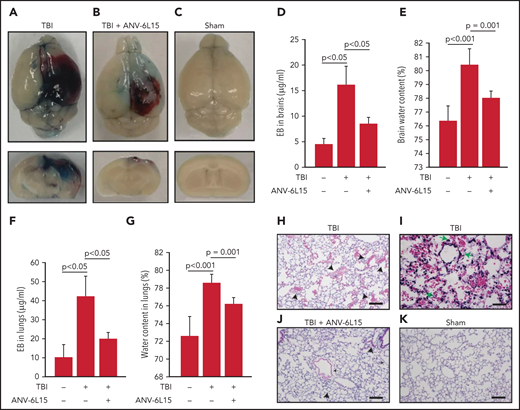
ANV-6L15 reduced TBI-induced vascular permeability. (A-C) Topical and coronal views of representative mouse brains collected 6 hours after TBI show hematoma (dark red) and vascular permeability (blue), both of which were reduced in TBI mice receiving ANV-6L15 and not observed in sham mice. (D) Summary of Evans blue tests from multiple experiments (n = 8 mice per group, 1-way ANOVA on ranks). (E) Brain-water content of TBI mice receiving either ANV-6L15 or control, and of sham mice (n = 8 mice per group, 1-way ANOVA). Lungs from these mice were examined for vascular leakage (F) and tissue edema (G) (n = 8 mice per group, one-way ANOVA). Representative H&E stains of lung sections show red cell accumulation in the alveolar spaces of TBI mice (H) (arrow: RBCs, scale bar, 50) and capillary dilation (I) (asterisk: RBC in alveolar space, green arrows: dilated capillaries; scale bar, 200 µm). The acute lung injury was drastically reduced in TBI mice receiving ANV-6L15 (J) (arrowhead: microbleed, arrow: interstitial edema; scale bar, 50 µm) and was not observed in sham mice (K) (scale bar, 50 µm).
ANV-6L15 reduced TBI-induced vascular permeability. (A-C) Topical and coronal views of representative mouse brains collected 6 hours after TBI show hematoma (dark red) and vascular permeability (blue), both of which were reduced in TBI mice receiving ANV-6L15 and not observed in sham mice. (D) Summary of Evans blue tests from multiple experiments (n = 8 mice per group, 1-way ANOVA on ranks). (E) Brain-water content of TBI mice receiving either ANV-6L15 or control, and of sham mice (n = 8 mice per group, 1-way ANOVA). Lungs from these mice were examined for vascular leakage (F) and tissue edema (G) (n = 8 mice per group, one-way ANOVA). Representative H&E stains of lung sections show red cell accumulation in the alveolar spaces of TBI mice (H) (arrow: RBCs, scale bar, 50) and capillary dilation (I) (asterisk: RBC in alveolar space, green arrows: dilated capillaries; scale bar, 200 µm). The acute lung injury was drastically reduced in TBI mice receiving ANV-6L15 (J) (arrowhead: microbleed, arrow: interstitial edema; scale bar, 50 µm) and was not observed in sham mice (K) (scale bar, 50 µm).
ANV-6L15 reduced TBI-IC
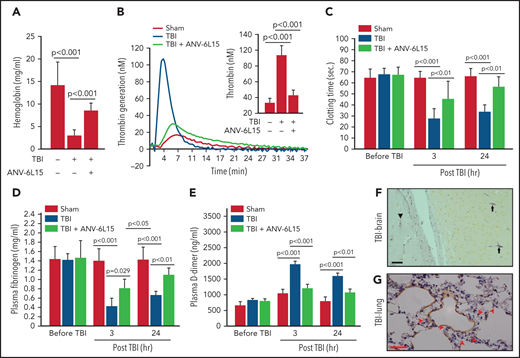
The histopathological data presented in Figures 1 and 2 suggest that ANV-6L15 reduced the TBI-induced hypercoagulable state and the resultant coagulopathy. Consistent with this notion, the TBI mice had significantly shortened tail bleeding 1 hour after injury, which was partially reversed by ANV-6L15 without prolonging the tail bleeding, measured by levels of hemoglobin (Figure 3A) and erythrocyte counts (supplemental Figure 4). This TBI-induced hypercoagulable state was also indicated by enhanced thrombin generation (Figure 3B), shortened clotting time (Figure 3C), reduced plasma fibrinogen (Figure 3D), and increased plasma D-dimer (Figure 3E), all of which were reversed by ANV-6L15 given 30 minutes post-TBI. The hypercoagulable state was also indicated by vascular fibrin deposition, a hallmark of the intravascular coagulation, in the brains (Figure 3F) and lungs (Figure 3G). Fibrin-rich thrombi were sporadically detected in the pulmonary vasculature (supplemental Figure 5). Together, these results demonstrate that TBI induced acute hypercoagulable and hyperfibrinolytic states that were blocked by ANV-6L15.

ANV-6L15 blocked the TBI-induced hypercoagulable state. (A) TBI significantly shortened tail bleeding, measured 1 hour post-FPI, and this was partially reversed by ANV-6L15 (n = 7 mice per group, 1-way ANOVA). (B-E) The TBI-induced hypercoagulation and its blockage by ANV-6L15, as measured by thrombin generation (B) (graph: thrombin generation plots, bar-graph insert: peak-thrombin generation, n = 9 mice per group), clotting time (C), plasma fibrinogen (D), and D-dimer (E). The data in panels C-E was analyzed using 1-way ANOVA (n = 9 mice per group). (F-G) Representative images of intravascular fibrin deposition detected using PTAH stains in cerebral (F) (arrowhead: microvascular bleeding, arrow: fibrin; scale bar, 20 µm) and pulmonary interstitial microvessels (G) (red arrow: fibrin deposition; scale bar, 50 µm) of TBI mice (×20 objective).
ANV-6L15 blocked the TBI-induced hypercoagulable state. (A) TBI significantly shortened tail bleeding, measured 1 hour post-FPI, and this was partially reversed by ANV-6L15 (n = 7 mice per group, 1-way ANOVA). (B-E) The TBI-induced hypercoagulation and its blockage by ANV-6L15, as measured by thrombin generation (B) (graph: thrombin generation plots, bar-graph insert: peak-thrombin generation, n = 9 mice per group), clotting time (C), plasma fibrinogen (D), and D-dimer (E). The data in panels C-E was analyzed using 1-way ANOVA (n = 9 mice per group). (F-G) Representative images of intravascular fibrin deposition detected using PTAH stains in cerebral (F) (arrowhead: microvascular bleeding, arrow: fibrin; scale bar, 20 µm) and pulmonary interstitial microvessels (G) (red arrow: fibrin deposition; scale bar, 50 µm) of TBI mice (×20 objective).
When tested separately, EVs, but not EV-free plasma from TBI mice, promoted thrombin generation that was blocked by ANV-6L15 (Figure 4A). The effect of ANV-6L15 was reversed by an anti-ANV antibody (supplemental Figure 6A) and was not observed in TBI mice pretreated with lactadherin (supplemental Figure S6B), which removes EVs from circulation through phagocytosis,13 suggesting that ANV-6L15 blocked PS-driven coagulation on EVs. Consistent with this finding, more than 80% of the circulating EVs found in TBI mice expressed PS, as detected by ANV binding (Figure 4B). This EV-induced coagulation was further supported by the finding that BDEVs induced thrombin generation (Figure 4C) and shortened clotting time (supplemental Figure 7) in a dose-dependent fashion. The BDEV-induced thrombin generation was dose-dependently blocked by ANV-6L15 (Figure 4D). There were more circulating BDEVs than EVs from pEVs and eEVs, and ANV-6L15 significantly reduced the levels of all 3 major EVs (Figure 4E). The levels of leuEVs were also increased but were significantly lower than those of BDEVs, pEVs, and eEVs (Figure 4F).
![A hypercoagulable state was induced primarily by EVs in TBI mice and reversed by ANV-6L15. (A) Blood samples from experimental mice were processed to generate PPP, concentrated EVs, and EV-free plasma (EVFP) and tested for thrombin generation (n = 7 mice per group, 1-way ANOVA). (B) Plasma levels of EVs that bound ANV in TBI and sham mice (n = 10 mice per group, Student t test). (C) BDEVs promoted thrombin generation in a dose-dependent fashion. The graph shows thrombin generation profiles, and the bar-graph insert summarizes the results of 12 independent experiments (1-way ANOVA; *P < .01 vs normal plasma). (D) Thrombin generation induced by 3 × 104/µL of BDEVs was blocked by ANV-6L15 in a dose-dependent manner. The bar-graph insert summarizes the results of 12 independent experiments (1-way ANOVA; *P < .01 vs normal plasma). Plasma levels of EVs from the brains (BDEV, using glial cell EVs [GFAP+] as the surrogate marker), eEVs, and pEVs (E) as well as leuEVs (F) in sham mice and TBI mice receiving ANV-6L15 or an equal volume of the vehicle buffer (n = 7 mice per group, 1-way ANOVA).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/25/10.1182_blood.2021011310/5/m_bloodbld2021011310r1f4.png?Expires=1770001970&Signature=aVgQEO2O3bOmoB-WGaftYl31jhi7Gk3hI0kSzSNYwl0e69wjXXLfYRMXBg5c~cKt50F3QPNu4P5muDhOIHC4ZjlUuovBqlpP3XwAZzadJqNhLz8inHPqIg2Bo2YfzZ0IwTQb4Md~zJ9HCicBnGsA4R-98MKeiFnQefwRxv7KXUTczXXluP6sa1NwDOjvJoJGyzndAwglkL~aiUsTMXLxtmVRrFopdh-Dx6uuPfKHuP8mXk2ywwTwGZbXSo5~JhQpEBtqzHT4I76H8thuRtZVjz4SY1eaCo75A-IjOuMGb9fukX8H9cGL9ynvTY4WGK2OwbEFcyjuigr6mTws8pOniw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
A hypercoagulable state was induced primarily by EVs in TBI mice and reversed by ANV-6L15. (A) Blood samples from experimental mice were processed to generate PPP, concentrated EVs, and EV-free plasma (EVFP) and tested for thrombin generation (n = 7 mice per group, 1-way ANOVA). (B) Plasma levels of EVs that bound ANV in TBI and sham mice (n = 10 mice per group, Student t test). (C) BDEVs promoted thrombin generation in a dose-dependent fashion. The graph shows thrombin generation profiles, and the bar-graph insert summarizes the results of 12 independent experiments (1-way ANOVA; *P < .01 vs normal plasma). (D) Thrombin generation induced by 3 × 104/µL of BDEVs was blocked by ANV-6L15 in a dose-dependent manner. The bar-graph insert summarizes the results of 12 independent experiments (1-way ANOVA; *P < .01 vs normal plasma). Plasma levels of EVs from the brains (BDEV, using glial cell EVs [GFAP+] as the surrogate marker), eEVs, and pEVs (E) as well as leuEVs (F) in sham mice and TBI mice receiving ANV-6L15 or an equal volume of the vehicle buffer (n = 7 mice per group, 1-way ANOVA).
A hypercoagulable state was induced primarily by EVs in TBI mice and reversed by ANV-6L15. (A) Blood samples from experimental mice were processed to generate PPP, concentrated EVs, and EV-free plasma (EVFP) and tested for thrombin generation (n = 7 mice per group, 1-way ANOVA). (B) Plasma levels of EVs that bound ANV in TBI and sham mice (n = 10 mice per group, Student t test). (C) BDEVs promoted thrombin generation in a dose-dependent fashion. The graph shows thrombin generation profiles, and the bar-graph insert summarizes the results of 12 independent experiments (1-way ANOVA; *P < .01 vs normal plasma). (D) Thrombin generation induced by 3 × 104/µL of BDEVs was blocked by ANV-6L15 in a dose-dependent manner. The bar-graph insert summarizes the results of 12 independent experiments (1-way ANOVA; *P < .01 vs normal plasma). Plasma levels of EVs from the brains (BDEV, using glial cell EVs [GFAP+] as the surrogate marker), eEVs, and pEVs (E) as well as leuEVs (F) in sham mice and TBI mice receiving ANV-6L15 or an equal volume of the vehicle buffer (n = 7 mice per group, 1-way ANOVA).
ANV-6L15-bound EVs to block the formation of extrinsic tenase complex
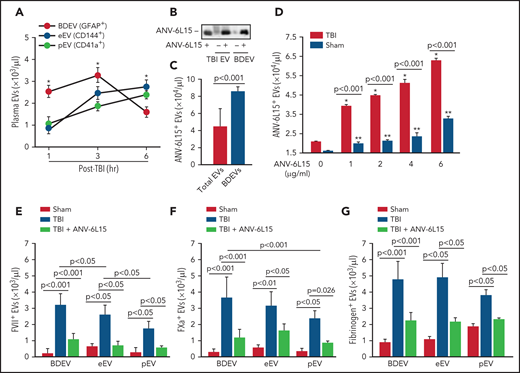
BDEVs were detected primarily in peripheral blood in the early hours after TBI and reduced over time, whereas the levels of eEVs and pEVs increased gradually during the same period (Figure 5A). ANV-6L15 infused into TBI mice was detected on EVs circulating in TBI mice, as well as on BDEVs made in vitro that were incubated with ANV-6L15 (Figure 5B). However, ANV-6L15 bound to BDEVs at a higher level than it did to heterogeneous EVs concentrated from TBI mice (Figure 5C). When incubated with EVs concentrated from TBI and sham mice in vitro, ANV-6L15 bound more EVs from TBI mice than from sham mice (Figure 5D) and also blocked ANV-binding to EVs in a dose-dependent manner (supplemental Figure 8), suggesting that ANV-6L15-bound PS exposed on EVs. Tissue factor was detected primarily on BDEVs12 and also on eEVs and leuEVs, albeit at much lower levels, and it was only detected minimally on pEVs; however, TF expression was not affected by ANV-6L15 (supplemental Figures 9), suggesting that TF expression was intrinsic and not acquired through PS. We detected FVII (Figure 5E), FXa (Figure 5F), and fibrinogen (Figure 5G) on EVs from TBI mice, with FVII and FXa at higher levels on BDEVs (GFAP+) than on eEVs (CD144+) or pEVs (CD41+). Together, these results suggest that the extrinsic tenase complex (TF, FVII, and Ca2++) was formed on PS-exposed EVs from TBI mice, especially on BDEVs, and its formation was blocked by ANV-6L15.
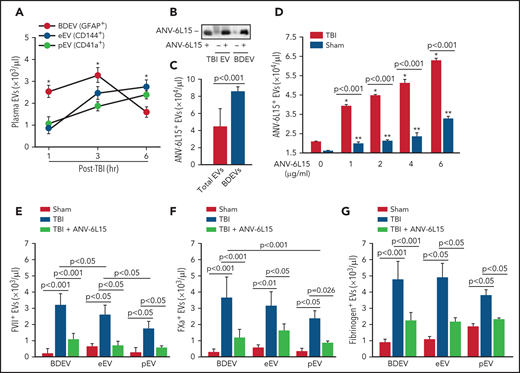
ANV-6L15 blocked the assembly of extrinsic tenase complexes on EVs. (A) Time-dependent changes in the circulating EVs in peripheral blood samples from TBI mice (n = 9 mice per group, 1-way ANOVA; *P < .05; BDEVs vs pEVs and eEVs, no statistical difference between pEVs, and eEVs). (B) ANV-6L15-bound EVs concentrated from TBI mice and BDEVs made in vitro were detected by immunoblots (representative of 3 separate experiments). (C) ANV-6L15 bound more BDEVs than did heterogeneous EVs from TBI mice (n = 10 mice per group, Student t test). (D) ANV-6L15-bound EVs concentrated from TBI and sham mice in a dose-dependent fashion (detected by a FITC-anti-ANV antibody, n = 9 mice per group, 1-way ANOVA; *P < .001; **P < .01 vs untreated). (E-G) FVII (E), FXa (F), and fibrinogen (G) were detected on the surfaces of EVs from TBI mice, with the highest levels found on BDEVs. ANV-6L15 reduced the formation of the tenase complex (n = 9 mice per group, Kruskal-Wallis 1-way ANOVA on ranks).
ANV-6L15 blocked the assembly of extrinsic tenase complexes on EVs. (A) Time-dependent changes in the circulating EVs in peripheral blood samples from TBI mice (n = 9 mice per group, 1-way ANOVA; *P < .05; BDEVs vs pEVs and eEVs, no statistical difference between pEVs, and eEVs). (B) ANV-6L15-bound EVs concentrated from TBI mice and BDEVs made in vitro were detected by immunoblots (representative of 3 separate experiments). (C) ANV-6L15 bound more BDEVs than did heterogeneous EVs from TBI mice (n = 10 mice per group, Student t test). (D) ANV-6L15-bound EVs concentrated from TBI and sham mice in a dose-dependent fashion (detected by a FITC-anti-ANV antibody, n = 9 mice per group, 1-way ANOVA; *P < .001; **P < .01 vs untreated). (E-G) FVII (E), FXa (F), and fibrinogen (G) were detected on the surfaces of EVs from TBI mice, with the highest levels found on BDEVs. ANV-6L15 reduced the formation of the tenase complex (n = 9 mice per group, Kruskal-Wallis 1-way ANOVA on ranks).
The detection of various levels of the extrinsic tenase complex on EVs from different parental cells led us to quantify anionic phospholipids, which bind ANV-6L15 and are required for the rapid initiation and propagation of coagulation. We found that EVs from TBI mice contained far more PS and had a significantly higher PS-to-PC ratio than those from sham mice (Table 2). BDEVs contained more PS than eEVs or pEVs did, even though the PS-to-PC ratios were comparable among the 3 types of EVs. In contrast, EV-free plasma contained a residual amount of PS but had a comparable PC level to that of EVs, resulting in an extremely low PS-to-PC ratio. Together, these results suggest that EVs from different parental cells express differential levels of PS, with BDEVs at the highest level; they also suggest that EVs, especially BDEVs, are the preferred surface to form the extrinsic tenase complex and are preferentially targeted by ANV-6L15.
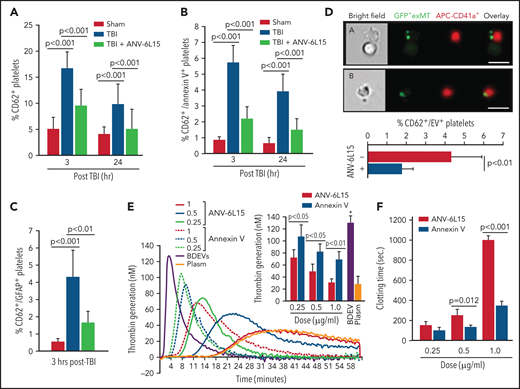
TBI mice developed significant thrombocytopenia (supplemental Table 3) and had increasing levels of activated (CD62p+) platelets (Figure 6A), suggesting consumptive thrombocytopenia. The severity of thrombocytopenia and the level of activated platelets were reduced in the TBI mice receiving ANV-6L15. More than 15% of circulating platelets in TBI mice expressed the α-granule protein CD62p (Figure 6A), but only 37% of these CD62p+ platelets (∼5% of the total platelets) expressed PS recognized by ANV (Figure 6B). The ANV binding on activated platelets was significantly less than that on pEVs, which had lower ANV-binding than eEVs or BDEVs (supplemental Figure 10). We also detected the platelet-BDEV complex in TBI mice (Figure 6C), suggesting that ANV also bound platelet-bound BDEVs. A previous study reported that platelets express an NSE subunit.30 However, neither NSE nor GFAP was detected on the surface of resting or activated platelets and endothelial cells as well as their EVs in this study (supplemental Figure 11), suggesting that the GFAP that was detected on platelets came from BDEVs. To validate the role of anionic phospholipid on the BDEV-platelet interaction and to reduce the heterogeneity of membrane EVs, we extracted exMTs from GFP+ mice and infused them into noninjured mice. These exMTs, which account for more than 50% of the EVs found in the circulation of TBI mice,21 formed complexes with platelets that were detected quickly after infusion (Figure 6D). BDEVs and exMTs bound platelets through PS and cardiolipin, respectively, mediated by the lipid receptor CD36 on platelets,25 and they could activate platelets through differential pathways.12,21 Levels of BDEV-platelet and exMT-platelet complexes were reduced in the TBI mice receiving ANV-6L15.
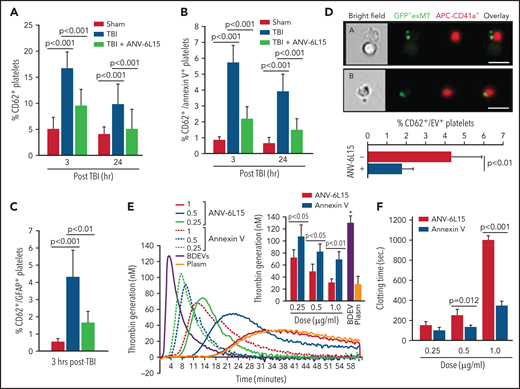
The severity of thrombocytopenia and the level of activated platelets were reduced in the TBI mice receiving ANV-6L15. Circulating platelets that expressed CD62p (A) and those that expressed both CD62p and PS (B) (n = 9 mice per group, 1-way ANOVA). (C) Platelets formed a complex with BDEVs and its blockage by ANV-6L15 (n = 9 mice per group, one-way ANOVA). (D) Platelets formed a complex with exMT in the absence and presence of 1 µg/ml of ANV-6L15, as detected by image flow cytometry. Top panel: representative images of GFP-exMT-platelet complexes (scale bar, 5 µm). Bottom panel: summary of 12 independent experiments (Student t test). (E) Thrombin generation induced by 3.5 × 103/µL of BDEVs in the presence of increasing concentrations of either ANV-6L15 or ANV. Graph: representative images. Insert: summary of 26 independent experiments (1-way ANOVA). (F) Clotting time induced by 6 × 103/µL of BDEVs in the presence of either ANV-6L15 or ANV (n = 13 mice per group, 1-way ANOVA). To be consistent with other experiments, ANV-6L15 was compared with ANV by weight (µg/mL), not by molar concentrations. Because of a smaller molecular mass, the results suggest that ANV was even less efficacious than ANV-6L15 in blocking thrombin generation.
The severity of thrombocytopenia and the level of activated platelets were reduced in the TBI mice receiving ANV-6L15. Circulating platelets that expressed CD62p (A) and those that expressed both CD62p and PS (B) (n = 9 mice per group, 1-way ANOVA). (C) Platelets formed a complex with BDEVs and its blockage by ANV-6L15 (n = 9 mice per group, one-way ANOVA). (D) Platelets formed a complex with exMT in the absence and presence of 1 µg/ml of ANV-6L15, as detected by image flow cytometry. Top panel: representative images of GFP-exMT-platelet complexes (scale bar, 5 µm). Bottom panel: summary of 12 independent experiments (Student t test). (E) Thrombin generation induced by 3.5 × 103/µL of BDEVs in the presence of increasing concentrations of either ANV-6L15 or ANV. Graph: representative images. Insert: summary of 26 independent experiments (1-way ANOVA). (F) Clotting time induced by 6 × 103/µL of BDEVs in the presence of either ANV-6L15 or ANV (n = 13 mice per group, 1-way ANOVA). To be consistent with other experiments, ANV-6L15 was compared with ANV by weight (µg/mL), not by molar concentrations. Because of a smaller molecular mass, the results suggest that ANV was even less efficacious than ANV-6L15 in blocking thrombin generation.
ANV-6L15 was more effective than ANV
Because ANV-6L15 contains ANV, which is a PS-binding protein31 and possesses antithrombotic activity,32,33 we compared ANV-6L15 to ANV for anticoagulant activity. At comparable doses, ANV-6L15 was significantly more efficacious than ANV in reducing EV-induced thrombin generation (Figure 6E) and prolonging clotting time (Figure 6F). It was also more efficacious than ANV in prolonging the tail bleeding of noninjured mice infused with BDEVs (supplemental Figure 12). These results are consistent with a previous report that ANV displaces coagulation factors incompletely, with residual surface-bound FXa, Va, and prothrombin to produce thrombin.34 They further suggest that ANV-6L15 is more effective because it not only blocks PS exposure but also has an intrinsic anticoagulant activity.
Discussion
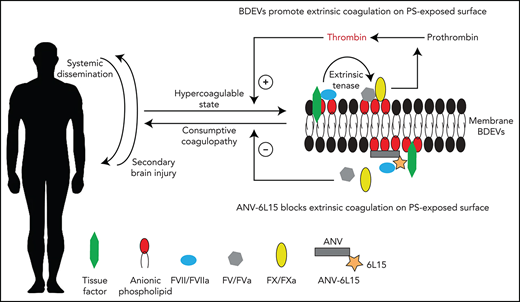
We presented data from a study designed to test the hypothesis that anticoagulation targeting membrane-bound anionic phospholipids prevents TBI-induced coagulopathy and improves the outcomes of TBI. This hypothesis was developed on the basis of our findings that severe TBI induces a consumptive coagulopathy developed from a hypercoagulable state, and that this consumptive TBI-IC is caused by anionic phospholipid-expressing EVs released from injured brains (BDEVs), including exMTs, and propagated by EVs from activated endothelial cells and platelets.12,13,21 We made the following novel and important observations.
First, ANV-6L15 given as a bolus dose 30 minutes post-TBI reduced subdural and intracerebral hematoma, improved neurological functions, and prevented the death of mice subjected to severe FPI (Figure 1). It protected the TBI mice by decreasing vascular permeability and tissue edema (Figure 2), preventing the injury-induced hypercoagulable state (Figure 3) and reducing circulating EVs (Figure 4) without significantly increasing the risk of bleeding. The finding that the anticoagulant ANV-6L15 prevented injury-induced bleeding is counterintuitive but consistent with the consumptive nature of TBI-IC. Equally important, our results show that a single physical impact applied to a small area of the brain through the dura mater induced not only cerebral injury but also injury to the lungs (Figure 2) and the heart (Table 1). This finding supports the clinical observation that cardiac and pulmonary pathologies develop frequently in patients with severe TBI; the former includes bradycardia, and the latter includes acute lung injury, acute respiratory distress syndrome, pneumonia, pleural effusions, pulmonary edema, and pulmonary thromboemboli.29,35 Our studies implicated BDEVs as the initiating, disseminating, and causal factors of the TBI-induced systemic endotheliopathy and coagulopathy.12,13
Second, we found that the extrinsic tenase complex36,37 was formed on EVs from brain cells, endothelial cells, and platelets during acute TBI, with the highest levels found on BDEVs (Figure 5). These results provide direct evidence that EVs serve as circulating mini platforms on which extrinsic coagulation is initiated and propagated, resulting in systemic consumption of coagulation factors and platelets that leads to local hemostatic defects at the site of the vascular injury (consumptive coagulopathy). ANV-6L15 blocked this TBI-induced and EV-driven hypercoagulable state, thus preventing secondary bleeding by delivering and localizing anticoagulant activity to membranes with exposed PS, which is a key component of the extrinsic tenase complex.
Third, the membrane-embedded PS accelerates the extrinsic coagulation by facilitating FVII binding to TF to proteolytically activate FX.38,39 We have previously shown that the membrane-bound anionic phospholipid cardiolipin is 1600 times as active as purified cardiolipin micelles in promoting coagulation.21 Here, we further show that EVs from TBI mice had significantly higher levels of PS and much higher PS-to-PC ratios than those from sham mice (Table 2). These high levels of PS could be attributed to BDEVs because they contained far more PS than eEVs and pEVs did, even though the PS-to-PC ratios were comparable among the 3 types of EVs (Table 2).12,40 The membrane-bound PS facilitates the formation of the tenase complex (Figure 5) and also makes BDEVs the preferential targets for ANV-6L15 because ANV binds PS in a density-dependent manner and each ANV monomer covalently binds 4 to 5 PS molecules,41,42 thus preferentially binding to membrane with a high PS density. This binding avidity is necessary because ANV has a very low affinity for individual PS molecules.43,44 For the same reason, ANV-6L15 preferentially binds EVs over their parental cells because the former has a high PS density. Furthermore, it has been shown that platelet-derived EVs are 50 to 100 times as procoagulant as activated platelets because they bind far more FX than platelets do.36,45 The tenase is also formed more efficiently when FVII binds the membrane-anchored TF,46 and it is most active in cleaving the membrane-bound FX.47 The BDEVs and exMTs were capable of not only activating platelets to express procoagulant activity and to generate pEVs during acute TBI but also bringing additional anionic phospholipids to the surface of these cells. Our data further shows that ANV-6L15 binds not only PS but also cardiolipin exposed on exMTs, which can account for more than 50% of ANV-bound EVs found in the circulation of TBI mice.21
Finally, ANV-6L15 acts distinctively from lactadherin13 to improve outcomes of TBI. As a scavenging factor, lactadherin needs PS-binding domains (C1C2) and an RGD sequence, which binds integrins (primarily β5) on the surface of macrophages and monocytes, in order to bring EVs to these cells to accelerate phagocytosis and EV clearance. In contrast, ANV-6L15 binds PS-exposed membrane surfaces to block the formation of the extrinsic tenase complex. However, we did notice that the ANV-6L15-treated mice had reduced numbers of circulating EVs relative to the control mice (Figure 4E-F), providing additional therapeutic benefits. How ANV-6L15 reduced circulating EVs remains to be investigated, but it is unlikely that this was caused by accelerating EV clearance because neither ANV nor ANV-6L15 contains the RGD sequence that is necessary for coupling EVs to macrophages and monocytes to facilitate phagocytosis. One possibility is that the ANV linked to ANV-6L15 stabilizes the cell membrane by changing the organization of membrane lipids, as previously reported,48-50 thus reducing the rate of cellular microvesiculation.
We chose 3 hours post-TBI as the time of key outcome measurements because circulating BDEVs reached their peak levels at this time, as shown in Figure 5A and our early report.12 This time point is clinically relevant for intervention, but it would not allow us to detect changes occurring at earlier time points. We are also unable to distinguish the impact of ANV-6L15 on PS-expressing EVs and on activated platelets and ECs, which also express PS. However, ANV-6L15 is likely to act on EVs preferentially because the binding of ANV to PS is density dependent, and EVs expressed far more PS than their parental cells (Table 2).
In summary, we found that ANV-6L15 targeted the extrinsic coagulation that was initiated and propagated on PS-exposed EVs and activated or injured cells to protect mice from TBI-induced cerebral and systemic injury. We showed differential PS and PC profiles of EVs from the brains, endothelial cells, and platelets for the first time, and more importantly, we linked these profiles to the formation of the extrinsic tenase complex. These findings delineate an important mechanism of TBI-induced endotheliopathy and consumptive coagulopathy and identify a new targeted therapeutic approach.
Acknowledgments
The authors thank Futoshi Shibasai and Hiroshi Shitara of Laboratory for Transgenic Technology, Animal Research Division, Tokyo Metropolitan Institute of Medical Science, as well as RIKEN BRC Japan for providing GFP+ mice. This study is supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL152200 and HL154250 (J.-f.D.) and Natural Science Foundation of China grants 81720108015 and 81930031 (J.Z.), and 81871919 (M.L.).
Authorship
Contribution: X.D. conducted experiments, analyzed data, and wrote the manuscript; W.L. performed histopathology review and wrote the manuscript; Y.S. performed mass spectrometry experiments and analyzed the data; K.H. performed experiments; M.Y., Y.Z., and Z.Z. performed experiments and analyzed data; X.W. designed flow cytometry experiments, analyzed data, and wrote the manuscript; T.B. performed mouse MRI and analyzed data; A.L.K. performed mouse tissue pathology; T.-C.W. developed and characterized ANV-6L15; X.F. designed MS experiments, analyzed data, and wrote the manuscript; M.L. formulated hypothesis, designed the study, analyzed histological data, and wrote the manuscript; J.Z. formulated hypothesis, designed the study, and wrote manuscript; and J.-f.D. formulated hypothesis, designed the study, analyzed data, and wrote manuscript.
Conflict-of-interest disclosure: T.-C.W. is the proprietor of EVAS Therapeutics, LLC. The remaining authors declare no competing financial interests.
Correspondence: Jing-fei Dong, Bloodworks Research Institute, 1551 Eastlake Ave East, Seattle, WA; e-mail: jfdong@bloodworksnw.org; Jianning Zhang, Department of Neurosurgery, Tianjin Medical University General Hospital, Tianjin, China; e-mail: jianningzhang@hotmail.com; and Min Li, Institute of Pathology, School of Medical Sciences, Lanzhou University, Lanzhou, China; e-mail: minli@lzu.edu.cn.
For original data, please contact jfdong@bloodworksnw.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

![A hypercoagulable state was induced primarily by EVs in TBI mice and reversed by ANV-6L15. (A) Blood samples from experimental mice were processed to generate PPP, concentrated EVs, and EV-free plasma (EVFP) and tested for thrombin generation (n = 7 mice per group, 1-way ANOVA). (B) Plasma levels of EVs that bound ANV in TBI and sham mice (n = 10 mice per group, Student t test). (C) BDEVs promoted thrombin generation in a dose-dependent fashion. The graph shows thrombin generation profiles, and the bar-graph insert summarizes the results of 12 independent experiments (1-way ANOVA; *P < .01 vs normal plasma). (D) Thrombin generation induced by 3 × 104/µL of BDEVs was blocked by ANV-6L15 in a dose-dependent manner. The bar-graph insert summarizes the results of 12 independent experiments (1-way ANOVA; *P < .01 vs normal plasma). Plasma levels of EVs from the brains (BDEV, using glial cell EVs [GFAP+] as the surrogate marker), eEVs, and pEVs (E) as well as leuEVs (F) in sham mice and TBI mice receiving ANV-6L15 or an equal volume of the vehicle buffer (n = 7 mice per group, 1-way ANOVA).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/25/10.1182_blood.2021011310/5/m_bloodbld2021011310r1f4.png?Expires=1770524720&Signature=A41OKtj2~zXA7vaEZ-EZcVrnge~qiE00hik8P3Hm8vEgf7V4DWSRJKLx1NrQZT0mHIEFRQ3cuSaaLbkAXxCsdiGFEtrvRw87dHm8OihKMp29GQItVWFcC3mzgTwefnRFWEctz7mozQm0GNMW~2g7YItXZ7PPbFYQ~Hf9mqfK9HUB1X9YbPHhKUJiU92jWpOa5otljcniREk5b8LirjZ2CVhhl57cnZK7Xxy~FTXZXNfgO~4t2frLyQ1OwE208H4Nai2RCWIGGfEBlzrZSJu4CCcXwSI5jli7WCkYQVse38FIyzk1wBfIjxGjw1z5GydvzxO-oF8U-bf3sQ8oBeLsfg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)