Abstract
Neurologic complications of lymphoid cancer can be challenging to recognize and treat. The nervous system can be affected directly by hematogenous or local spread of lymphoma. Indirect neurologic effects of lymphoma include paraneoplastic syndromes and vascular complications. Lymphoma treatments can also cause neurologic complications. Early identification and treatment are crucial to stabilize or reverse neurologic deficits, prevent further nervous system injury, and optimize overall oncologic therapy. This article provides an overview of the different neurologic complications of lymphoma and its treatments, in addition to presenting case studies that emphasize commonly encountered clinical scenarios.
Neurologic complications in lymphoid cancers can manifest in several ways: (1) direct complications: involvement of the central or peripheral nervous system by lymphoma; (2) indirect complications: spinal cord compression, paraneoplastic syndromes (PNSs), vascular complications and paraprotein associated neuropathies; and (3) treatment-associated complications. Depending on the specific cause, patients can present with mild, self-limiting symptoms, or severe, disabling, and irreversible neurologic deficits that can be fatal. As immunotherapies are being increasingly used in patients with lymphoma, there has been an increase in referral of patients for neurologic symptoms and signs. Given that various conditions can lead to the same clinical presentation, it can be challenging to determine the exact etiology. Differentiation between presentations is necessary to guide treatment.
In this review, we provide an overview of the neurologic complications of Hodgkin (HL) and non-Hodgkin (NHL) lymphoma, describe cases, and highlight practical difficulties encountered in the recognition and management of these complications. We do not discuss primary central nervous system (CNS) lymphoma, as it is beyond the scope of this review.
Direct complications
CNS involvement
NHL, rarely, can involve the CNS concurrently at the time of initial diagnosis or at the time of relapse. The median time to development of CNS relapse is less than a year and can occur as isolated CNS disease or in conjunction with systemic relapse of disease. The overall incidence of CNS involvement is 2% to 7% and, although several risk factors including histologic subtype (Burkitt’s lymphoma and lymphoblastic lymphoma) have been identified, the CNS International Prognostic Index, composed of variables such as older age, poor performance status, advanced-stage (III/IV) disease, ≥1 extranodal sites, elevated serum lactate dehydrogenase, and involvement of specific organs, such as the adrenal glands and kidneys, can help stratify patients at risk.1 Primary testicular lymphomas are associated with a high incidence of CNS relapse.2 Biomarkers including, double-hit lymphomas characterized by MYC and BCL2 and/or BCL6 rearrangements and activated B cell, such as diffuse large B-cell lymphomas (DLBCLs), that are associated with a poor prognosis also increase the risk of CNS involvement.3,4 CNS relapse with HL is rare, with an incidence of <0.5% and associated Epstein-Barr virus.5,6 CNS involvement from systemic NHL or HL may include the brain, spinal cord, eye, leptomeninges (includes subarachnoid space and cerebrospinal fluid [CSF]) and dura in isolation or in combination. Contrast-enhanced magnetic resonance imaging (MRI) of the brain and spine, CSF analysis, and/or brain biopsy are typically recommended for diagnosis. Ophthalmologic evaluation including a slit-lamp examination is recommended for those with symptoms of possible intraocular involvement. High-dose methotrexate (HD-MTX)–based chemotherapy followed by thiotepa-based autologous transplant in patients with secondary CNS DLBCL (SCNSL) is associated with a complete response (CR) rate of 63% and a 2-year event-free survival of 51%, and this treatment should be considered in the appropriate transplant-eligible patient.7 Targeted agents such as ibrutinib and lenalidomide have demonstrated response rates of 60% to 75% in patients with SCNSL, although these responses are not durable, and the median progression-free survival is ∼6 months.8,9 Whenever possible and if available, we recommend consideration of enrollment of patients in clinical trials. CD19-directed CAR T-cell therapy, which has improved outcomes of patients with relapsed and refractory DLBCL, is a promising treatment in SCNSL, with responses observed in 4 of 8 patients treated with tisagenleucleucel in a case series. Clinical trials are ongoing.10
Peripheral nervous system involvement
NHL can involve the peripheral nerves and plexi, by either direct invasion or infiltration. Neurolymphomatosis can cause painful peripheral neuropathy or radiculopathy. Occasionally, patients present with mononeuropathies of the sciatic, radial, median, or even cranial nerves, leading to radicular pain, paresthesias, and weakness in the legs or arms or diplopia and facial weakness, respectively. Diagnosis can be made by MRI of the involved area and fluorodeoxyglucose-positron emission tomography (FDG-PET) which typically shows increased uptake. Treatment involves focal radiation or systemic chemotherapy. Given that this is a rare complication, it is important to consider in the differential diagnosis other unrelated causes of neuropathies such as medications; vitamin or nutrient deficiencies; metabolic and autoimmune conditions; and, in patients with a history of NHL or HL, paraneoplastic causes, or chemotherapy-induced neuropathy.
Case 1
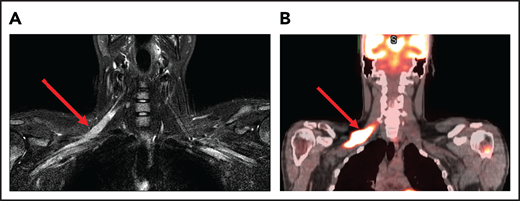
A 25-year-old man presented with fatigue with abdominal and bone pain, evaluation of which led to the diagnosis of a high-grade DLBCL with MYC and BCL2 rearrangements. Upon completion of treatment with 6 cycles of R-EPOCH (rituximab-etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) and prophylactic intrathecal (IT) methotrexate, he developed severe pain in his right shoulder and left leg, associated with numbness of his left foot. Over the next month, his symptoms progressed, and he developed worsening pain and weakness in the right arm and bilateral foot drop. He was then diagnosed with Parsonage-Turner syndrome, an idiopathic brachial plexopathy, and treated with methylprednisolone, which did not help. Subsequently, an MRI of the brachial plexus showed increased enhancement and FDG-PET scan showed increased uptake in the right brachial plexus and bilateral sciatic nerves (Figure 1). Finally, he was diagnosed with neurolymphomatosis. He responded to treatment with HD-MTX and cytarabine and made a full recovery.
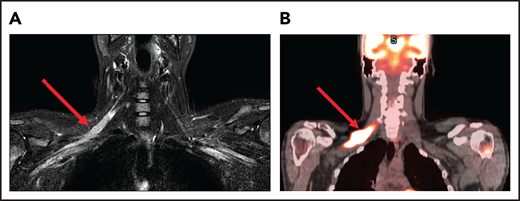
Brachial plexus MRI and FDG-PET scan of a patient with neurolymphomatosis. (A) An axial T1-weighted image after gadolinium contrast shows enhancement of the right brachial plexus. (B) FDG-PET scan shows an increase in FDG uptake in the right brachial plexus.
Brachial plexus MRI and FDG-PET scan of a patient with neurolymphomatosis. (A) An axial T1-weighted image after gadolinium contrast shows enhancement of the right brachial plexus. (B) FDG-PET scan shows an increase in FDG uptake in the right brachial plexus.
Case 1 highlights the fact that any patient with a history of an aggressive cancer, including lymphomas, who presents with new-onset neurologic symptoms or signs or change in quality, severity, or frequency of existing symptoms, must undergo a thorough evaluation to identify the etiology and rule out malignant involvement of the nervous system. Asymmetric neurological symptoms and signs are typically not related to chemotherapy-induced neuropathy.
Indirect complications
Spinal cord compression
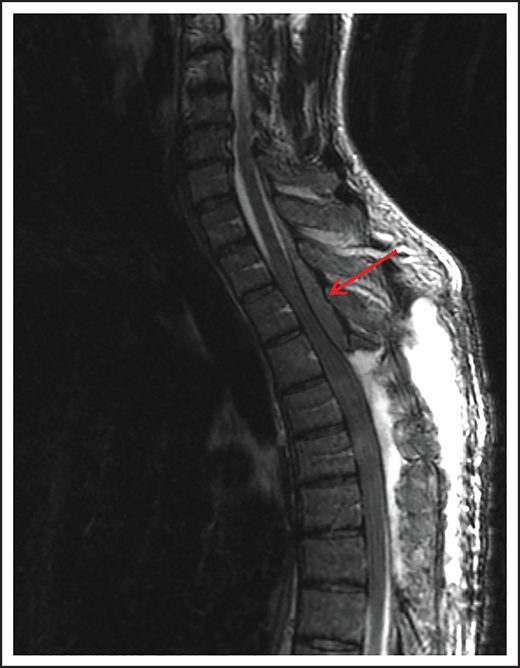
Spinal cord compression can cause severe and permanent neurologic deficits and must be diagnosed and treated emergently to reverse symptoms and prevent devastating sequelae. Epidural lesions caused by lymphoma involvement of the vertebral bodies of the spine or paravertebral lymph nodes can lead to spinal cord compression (Figure 2). In a retrospective study of 131 patients with primary bone lymphoma, one-third of the patients had involvement of the spine leading to cord compression in half of them.11 Most patients present with back pain, although neurologic symptoms including paralysis, sensory loss, and bladder and bowel dysfunction can evolve rapidly. Delayed diagnosis and treatment can result in permanent neurologic deficits. Diagnosis is typically made with MRI of the spine. Dexamethasone can improve neurologic symptoms and signs until definitive treatment, including chemotherapy and/or radiation can be instituted. Occasionally, patients may need neurosurgical intervention to maintain stability of the spine.
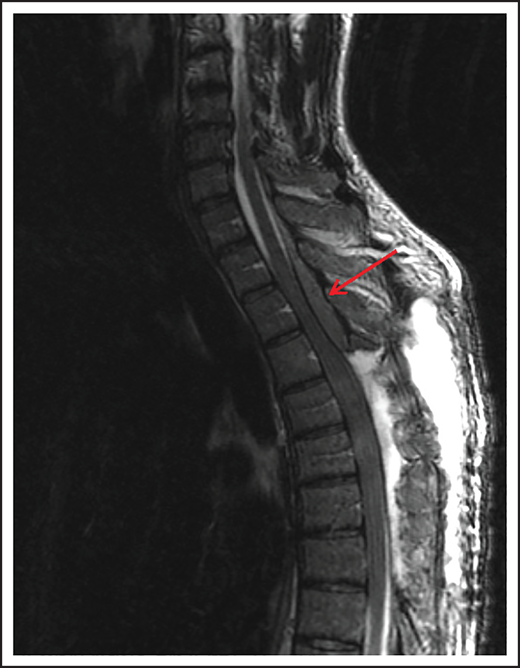
MRI of the cervical and thoracic spine of a patient with epidural spinal cord compression. A sagittal T2 image shows epidural involvement by NHL with compression of the underlying spinal cord.
MRI of the cervical and thoracic spine of a patient with epidural spinal cord compression. A sagittal T2 image shows epidural involvement by NHL with compression of the underlying spinal cord.
Paraneoplastic syndromes
Classic paraneoplastic syndromes (PNSs) are rare complications of lymphomas, and in fact, PNSs are associated with unique presentations with a higher incidence in HL than in NHL.12,13 Unlike the case with solid tumors, onconeural antibodies are rarely identified, with exceptions, such as paraneoplastic cerebellar degeneration (PCD) and limbic encephalitis, more commonly observed in HL than in NHL (Table 1). PCD presents with progressive symptoms of vertigo, truncal and limb ataxia, dysarthria, diplopia, and nystagmus. An MRI may initially be normal, and cerebellar atrophy occurs over time. Anti-Tr antibodies are found in the serum and CSF of patients.14 PCD from HL has a slightly better prognosis compared with PCD secondary to solid tumors, and improvement has been reported in patients <40 years of age upon treatment of the underlying HL. Paraneoplastic limbic encephalitis (Ophelia syndrome) presents with confusion, memory loss, hallucinations, and behavioral problems.15 A brain MRI may be unremarkable or associated with abnormalities in the bilateral medial temporal lobes, hippocampus, and amygdala. This rare syndrome is associated with an anti-mGLU5 (metabotropic glutamate receptor type 5) antibody. Often, patients have a full recovery after treatment of the underlying HL. Paraneoplastic sensorimotor neuropathies; neuronopathies or conditions resembling Guillain-Barré syndrome; or, more commonly, chronic inflammatory demyelinating polyradiculopathy (CIDP) present in patients with NHL with a subacute course and absence of onconeural antibodies. Sensory neuronopathies generally have a poor prognosis. Dermatomyositis is also observed in NHL and the presence of a p155 antibody is highly suggestive of an underlying malignancy.16 Outside of the classic syndrome of PCD, it can be challenging to make the diagnosis of PNSs in lymphoma, especially when there is no associated onconeural antibody. Suspected PNSs should trigger a diagnostic evaluation, as they typically present before lymphoma diagnosis, followed by prompt treatment of the same. In the absence of coexistent lymphoma, corticosteroids, IV immunoglobulin, plasma exchange, cyclophosphamide and rituximab are treatment options based on the specific type of PNS.
Vascular complications
Primary angiitis of the CNS is a rare, noninfectious, granulomatous condition that affects the cerebral blood vessels. This effect can be seen in association with HL, either immediately before or after diagnosis. Because the histopathologic picture resembles an autoimmune condition and because of its association with lymphoma, it is often considered to be a PNS, although it could be related to a varicella-zoster virus infection.17 Patients present with headaches, confusion, seizures, and focal deficits. Rarely, the deficit can involve the spinal cord, causing transverse myelitis. An MRI often shows infarcts or leptomeningeal enhancement, cerebral angiography may demonstrate beading suggestive of vasculitis, and CSF may demonstrate pleocytosis, but these are nonspecific findings. A brain biopsy is necessary for confirmation and occasionally may also show Aβ amyloid angiopathy.18 Patients typically recover with corticosteroids and treatment of HL.
Intravascular lymphoma (IVL) is an aggressive extranodal NHL involving the lumens of small-vessels, particularly capillaries. Although any organ can be involved, the CNS is the most common, occurring in 42% of IVL cases, followed by the skin.19 Patients typically present with stroke like symptoms, fever, fatigue, and skin rashes. A brain MRI typically demonstrates infarcts that are not specific for any vascular territory, and this finding should alert the clinician to this diagnosis. Often skin and bone marrow biopsies are enough, but occasionally a brain biopsy can be required for histopathologic confirmation. Treatment options include HD-MTX and cytarabine in addition to R-CHOP (rituximab-cyclophosphamide, doxorubicin, vincristine, and prednisone).20 The risk of relapse is high and prognosis is poor. The CNS is less likely to be involved in the hemophagocytosis variant of IVL in Asian patients.
Paraprotein-associated neuropathies
Peripheral neuropathy is the most common neurologic complication of paraproteinemias including lymphoplasmacytic lymphoma with IgM (Waldenström’s macroglobulinemia), monoclonal gammopathy of unknown significance, multiple myeloma, amyloid light chain amyloidosis, and POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome. Most of these polyneuropathies are associated with IgM paraprotein, up to 70% of which are related to the anti-myelin–associated glycoprotein (MAG) antibody.21,22 The peripheral neuropathy may present as a distal large-fiber sensory neuropathy; multiple mononeuropathies; or painful, length-dependent sensory, motor, or sensorimotor neuropathies that may be associated with autonomic dysfunction. POEMS syndrome may present with CIDP. Patients may present with progressive symmetric paresthesias, sensory impairment, pain, weakness, and often, tremor. Electrophysiologic studies can confirm neuropathy. High titers of anti-MAG antibodies can be detected in serum in untreated patients. A nerve biopsy may be necessary. Treatment of the underlying disease with rituximab monotherapy or in combination with chemotherapy may result in improvement of symptoms and reduction or normalization of anti-MAG antibody titers.23 Corticosteroids, IV immunoglobulin, and plasma exchange have been tried with varying success in patients with CIDP.
Treatment-related complications
Neurologic complications can result from the various treatments of NHL and HL, including chemotherapy, radiation, autologous stem cell transplant (SCT), allogeneic SCT, targeted therapies, and immunotherapy. Neurologic side effects from traditional chemotherapy are common, nonhematologic, dose-limiting toxicities in oncology.24 With the advent of targeted therapy and immunotherapy, specific targeting of tumors can occur that may lead to reduction in off-target side effects such as myelosuppression. However, some newer therapies have unique neurotoxicities, and their early recognition is critical. It is important to note that treatment-related neurotoxicity is a diagnosis of exclusion, and other etiologies should be considered first. Comprehensive diagnostic evaluation is warranted, including ruling out nervous system involvement by lymphoma as described earlier. Conditions such as headaches, strokes, peripheral neuropathies, and Guillain-Barré syndrome, all of which occur in patients without cancer, may develop in patients with lymphoma, and it is necessary to determine whether these are triggered by the lymphoma or its treatment or are completely unrelated. Several factors can affect the incidence and severity of the neurotoxicity. These include age, dose of drug and route of administration, concurrent renal or liver dysfunction, combined modality treatments, and preexisting neurologic conditions. For example, an older adult (>60 years of age) is at a higher risk of long-term neurocognitive decline from combination IV methotrexate and whole-brain radiation than a younger patient.25 Underlying conditions can predispose patients to early or exaggerated side effects, depending on the type of treatment involved. For example, a patient with an inherited motor and sensory neuropathy such as Charcot-Marie-Tooth disease is at a higher risk of chemotherapy-induced peripheral neuropathy from antimicrotubule drugs such as vincristine.26 A patient with a history of seizure disorder may be at a higher risk of seizures from treatment with high-dose busulfan or CD19 CAR T-cell therapy. For this reason, a detailed neurologic history and baseline neurologic examination are essential before starting treatment.
Lymphoma treatment can affect any part of the nervous system including the central and peripheral nervous systems (Table 2). Neurotoxicity in the CNS can manifest with nonspecific, nonlocalizing symptoms and signs such as headaches, confusion, seizure, or loss of consciousness which, in turn, can be manifestations of different underlying neurologic conditions such as meningitis, encephalitis, cerebral infarction, or intracerebral hemorrhage. Localizing or focal deficits such as weakness, aphasia, visual loss or seizures or can also be observed with these underlying neurological conditions. Patients with myelopathy may present with Lhermitte’s sign which is an electric shock–like sensation throughout the spine, limbs, and occasionally the trunk, triggered by bending the neck. Peripheral nervous system toxicity can present as a sensory or motor neuropathy, ascending polyradiculopathy like Guillain-Barré syndrome, myasthenia gravis from involvement of the neuromuscular junction or myositis. In general, diagnosing neurologic complications of lymphoma treatment is challenging, as the same set of symptoms and signs may result from direct complications from lymphoma, indirect (infectious, vascular, etc) complications of lymphoma, or a PNS. Once a treatment-related neurotoxicity is recognized, the determination must be made of whether the treatment should be discontinued or the side effect is dose dependent and dose adjustment can be made. In the following sections common therapeutics are described that are used to treat lymphoid cancers that have a propensity to cause neurologic complications.
Immunotherapies
Immune checkpoint inhibitors are approved treatments for HL (pembrolizumab and nivolumab) and primary mediastinal B-cell lymphoma (pembrolizumab). The incidence of immune-related neurologic toxicities from anti-PD1 antibodies is 6%.27,28 Headache and peripheral sensory neuropathy are more common manifestations, although patients treated with these therapies can develop a wide range of neurologic symptoms (Table 3). Immune-mediated encephalitis can present with acute onset of confusion, depressed level of consciousness, or personality change. Hypophysitis may also occur as a complication with acute onset of headaches, fatigue, and generalized weakness, in addition to endocrine abnormalities. Cerebellar toxicity can present with ataxia, nystagmus, and vertigo. Symptoms typically occur acutely or subacutely within 3 months of initiation of treatment, but can occur as early as after the first dose of treatment.29 Brain MRI and CSF should be obtained to rule out malignant spread, infection, and toxic-metabolic causes of altered mental status. Therapy should be discontinued in patients with grade 2 or higher neurotoxicity, and corticosteroids should be instituted rapidly to prevent fatal outcomes. Empiric antibiotic or antiviral coverage for encephalitis should be considered until an infectious etiology is excluded.27 Checkpoint inhibitors can also lead to polyradiculopathy or Guillain-Barré syndrome with facial diplegia, dysphagia, sensory loss, paresthesias, weakness, and areflexia. CSF can demonstrate a cytoalbuminologic dissociation (high protein with few cells), and demyelinating polyneuropathy is detected on a nerve conduction study. Corticosteroids and IV immunoglobulin are advised. Plasma exchange may also be considered for severe cases. Recovery may be full or incomplete; in the latter case, patients are left with residual neurologic deficits. Myositis can involve various muscle groups and typically causes proximal limb weakness, muscle pain, dysarthria, dysphagia, ptosis, diplopia, and occasionally, dyspnea from diaphragmatic weakness.30,31 Serum creatine kinase is elevated, and muscle biopsy may be needed for diagnosis, typically demonstrating necrotic and inflammatory changes. Corticosteroids and immune checkpoint discontinuation may allow for complete recovery, although prolonged morbidity and fatal cases have been described.31 Approximately 60% of patients with myasthenia are found to have acetylcholine receptor antibodies; and, despite aggressive treatment with pyridostigmine, corticosteroids, IVIG and plasmapheresis, this condition can be fatal in up to one-third of patients.32 Myasthenia gravis and myositis can occasionally occur at the same time, and both can overlap with myocarditis. For this reason, when one of these conditions is suspected, evaluation for the others is recommended.33 The risk of retreatment with an immune checkpoint inhibitor is not well defined and such a strategy should be considered on a case-by-case basis taking into account the severity/grade of the neurotoxicity, degree of recovery, any residual neurologic deficits, and the status of the lymphoma including alternative options.
Case 2
A 66-year-old man with systemic and CNS DLBCL was treated with R-CHOP and HD-MTX followed by an autologous SCT. Three months after transplant, the lymphoma relapsed in the ethmoid sinus. He underwent CD-19–directed CAR T-cell therapy and on postinfusion day 2, he developed headaches and mild aphasia followed by confusion. A noncontrast head CT was unremarkable, and an electroencephalogram did not show any epileptiform discharges. He responded to corticosteroid therapy and was discharged home in 9 days. He remained in remission more than a year later.
CAR T-cell therapy directed toward CD19 is approved for relapsed/refractory DLBCL, mantle cell lymphoma, and B-cell acute lymphoblastic leukemia and can cause immune effector cell–associated neurologic syndrome (ICANS) in association with cytokine-release syndrome.34 Acute encephalopathy with delirium and aphasia are frequently observed within the first 7 days of infusion, as in Case 2.35 Other symptoms may include headaches, hallucinations, tremors, and myoclonus. Often symptoms peak and then resolve within the next few days to weeks.36 Rarely, the syndrome can rapidly progress to generalized seizures, coma, and death. Brain MRI is typically unremarkable although occasionally T2 (FLAIR) fluid-attenuated inversion recovery hyperintensity without restricted diffusion in deep brain regions is observed and in rare cases, cerebral edema has been reported.37 Most patients are treated with and respond to corticosteroids.38 Anti-IL6 receptor antibody, tocilizumab approved for use in cytokine release syndrome, is not effective in ICANS and in fact may be associated with worsening neurotoxicity.39,40 There is preclinical evidence that the IL1 receptor antagonist anakinra prevents neurotoxicity, and clinical trials are ongoing.41 For all patients scheduled for CAR T-cell therapy, a baseline neurologic examination and ICANS grading scale should be completed on admission with frequent monitoring during the hospital course.
Monoclonal antibodies
Brentuximab vedotin is a CD30-specific antibody drug conjugate approved for relapsed/refractory HL and anaplastic large-cell lymphoma, and its most common neurologic side effect is peripheral neuropathy. A sensory neuropathy characterized by numbness, paresthesias, and burning pain is common, observed in 40% to 69% of patients, although patients can also present with motor neuropathy.42,43 Most patients present with grade 2 neuropathy, but some develop severe symptoms requiring temporary or permanent discontinuation of the drug. In case of a temporary hold, it is advised that brentuximab vedotin be readministered at a lower dose once symptoms are grade 1 or less. Severe pain from peripheral neuropathy can be managed by administering gabapentin, pregabalin, or duloxetine. These drugs are not typically recommended in the absence of pain, as they do not help to alleviate numbness, tingling, or paresthesias. Physical therapy may be helpful in patients with weakness from motor neuropathy. It is noteworthy that many patients receiving brentuximab vedotin may have had prior exposure to other drugs associated with peripheral nerve toxicity, such as vinca alkaloids and cisplatin, and this prior exposure may increase the risk of neurotoxicity. In most cases, symptoms resolve after completion of treatment.
Rituximab, an anti-CD20 antibody, and alemtuzumab, an anti-CD52 antibody, can rarely cause reactivation of latent JC virus infection leading to progressive multifocal leukoencephalopathy (PML). Many patients present with altered mental status, although some may have motor deficits, aphasia, or visual loss. Diagnosis is made by MRI and confirmed by CSF polymerase chain reaction for JC virus or brain biopsy.44,45 There is no standard treatment for PML, and most cases are fatal. Mefloquine, mirtazapine, and cytarabine have been tried. In a series of 8 patients with PML, immune checkpoint blockade with pembrolizumab resulted in clinical improvement or stabilization, as well as a reduction in CSF JC viral load in 5 of 8 patients, and is a promising therapy for this devastating neurologic complication.46 Case reports have indicated benefit from interleukin-2 and IL-7. Treatment with N-803, an IL-15 superagonist, resulted in a response in 1 patient with PML.47
Polatuzumab vedotin is an anti-CD79B monoclonal antibody conjugated to the microtubule disrupting agent monomethyl auristatin E, and it is implicated in peripheral sensory neuropathy in 44% to 67% of patients treated with this agent.48,49 Most of the patients report grade 1/2 neuropathy including paresthesias which typically resolved, although a higher grade neuropathy requiring treatment discontinuation has been reported.
Small-molecule inhibitors
Ibrutinib, a first-in-class BTK inhibitor is associated with bleeding and, rarely, intracerebral hemorrhage.50,51 Patients may present with headaches or focal neurologic deficits. If minimally symptomatic, then the drug can be restarted at a lower dose after resolution of hemorrhage and symptoms.
Lenalidomide, pomalidomide, and thalidomide, which are immunomodulatory drugs, can cause length-dependent axonal sensory neuropathy, typically several months after starting therapy.52 Nerve conduction test results are usually abnormal. Most patients report mild symptoms and do not require any treatment.
Case 3
A 69-year-old woman with a history of DLBCL treated with R-CHOP presented with diplopia 3 months after completion of therapy. On neurologic examination she had left abducens nerve (cranial nerve VI) palsy. A brain MRI with contrast did not demonstrate any abnormal leptomeningeal enhancement, infarct, or hemorrhage. Over the next week, her symptoms worsened. CSF tests did not reveal malignancy or other abnormality. After 10 days, the symptoms stabilized and over the next 2 months, her diplopia resolved.
Vinca alkaloids
Vincristine causes a dose-limiting, axonal, sensory, and motor neuropathy, and its incidence in patients with lymphomas is higher than when used in other cancers.53 Often patients present with paresthesias, wrist drop, or foot drop.54 Muscle cramps may be an initial sign of an impending neuropathy. Paresthesias can occur during treatment, but it is not uncommon for patients to develop symptoms months after completion of treatment, as seen in our case. Cranial neuropathies (as in case 3) including involvement of the facial nerve (facial weakness), oculomotor, and abducens nerves (diplopia, ptosis), optic nerve (visual loss), auditory nerve (hearing impairment), and recurrent laryngeal nerve (dysphagia) have been reported. Autonomic neuropathy presenting with orthostatic hypotension, constipation, and abdominal pain from ileus, and erectile dysfunction can occur. In general, the severity of the neuropathy increases with cumulative doses of vincristine and a dose-limit of 1.4 mg/m2 with a maximum dose of 2 mg per cycle is recommended.55 Concurrent administration of vincristine with the antifungal agents voriconazole and itraconazole is associated with enhanced neurotoxicity.56 Physical therapy is often necessary for recovery from motor neuropathies, and severe cases may not have a complete recovery. Gabapentin for prophylaxis or treatment is not beneficial.
Case 4
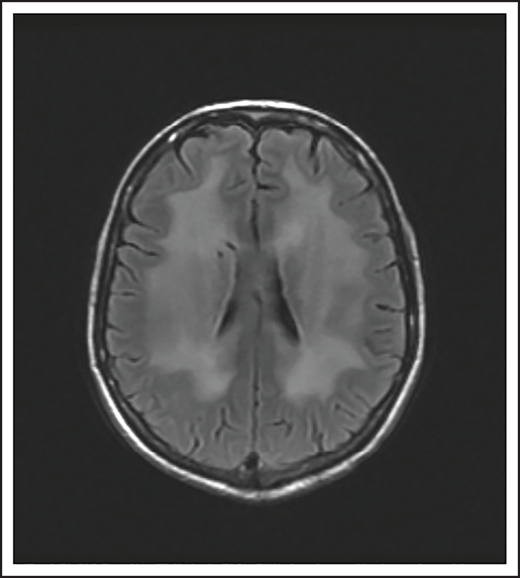
A 74-year-old man with a history of DLBCL treated with R-CHOP presented with progressive confusion and right-side weakness. A brain MRI demonstrated a contrast-enhancing lesion in the corpus callosum with extension into the left parietal region. A biopsy of the enhancing lesion confirmed a diagnosis of DLBCL. No other sites of disease were identified, and he was diagnosed with isolated CNS relapse. He was treated with HD-MTX, rituximab, and temozolomide for 4 cycles, and he achieved a complete radiographic response. Three years later, he presented with progressive memory loss, urinary incontinence, and gait ataxia. A brain MRI demonstrated periventricular white matter T2/FLAIR hyperintensities (Figure 3). Evaluation for toxic and metabolic causes of his symptoms was negative. His diagnosis was MTX-induced leukoencephalopathy.
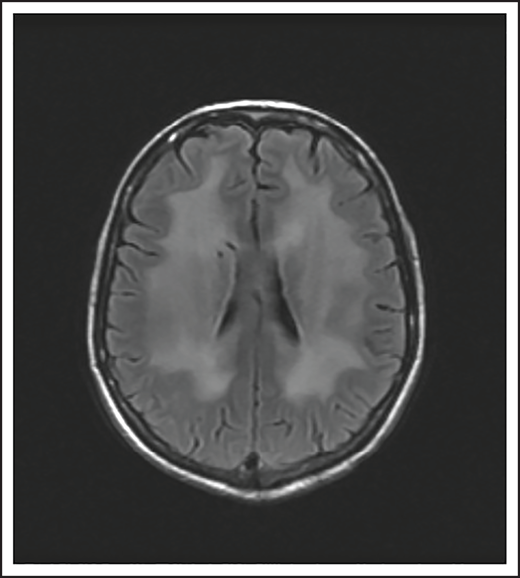
Brain MRI of a patient with leukoencephalopathy caused by use of HD-MTX. An axial T2 fluid attenuated inversion recovery (FLAIR) image shows periventricular hyperintense signal reflecting leukoencephalopathy.
Brain MRI of a patient with leukoencephalopathy caused by use of HD-MTX. An axial T2 fluid attenuated inversion recovery (FLAIR) image shows periventricular hyperintense signal reflecting leukoencephalopathy.
Antimetabolites
HD-MTX as well as IT MTX can cause acute as well as subacute encephalopathy.57,58 Acute encephalopathy is usually reversible. Subacute, or chronic, encephalopathy, as seen in case 4, is typically progressive, irreversible, and associated with high morbidity and mortality. It is a diagnosis of exclusion and as described in case 4, other causes must be excluded. The risk is higher with older age and when combined with whole-brain radiation therapy (WBRT).59
Cytarabine arabinoside (AraC) in high doses can result in an acute cerebellar syndrome characterized by incoordination, ataxia, gait imbalance, dysarthria, and nystagmus.60 Risk factors include preexisting liver or renal dysfunction, older age, and history of prior neurologic deficits. Typically, the cerebellar deficits resolve within 2 weeks of discontinuation of drug, although rarely, the syndrome can be permanent with cerebellar atrophy over time.
Case 5
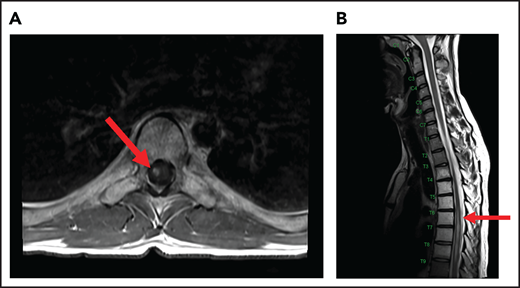
A 24-year-old man presented with stage IVB DLBCL and completed 6 cycles of R-CHOP, with FDG-PET after treatment showing resolution of all disease except in the mediastinum. After a biopsy of this residual FDG+-confirmed DLBCL, he received treatment with salvage chemotherapy with minimal response, followed by mediastinal involved field radiation therapy (IFRT) to a dose of 40 Gy plus a 6-Gy boost, after which he had a CR. Subsequently, he underwent autologous SCT followed by RIC allogeneic SCT. He remained in CR for 16 months, at which time he presented with 3 weeks of new right leg weakness, erectile dysfunction, and urinary hesitancy. The results of a brain MRI were unremarkable, but a spine MRI demonstrated spinal cord expansion and edema with multifocal enhancement within the prior radiation therapy portal from T5 to T9, demarcated by fatty marrow replacement between the T5 and T9 vertebral bodies (Figure 4). A diagnostic evaluation, including CSF analysis, was negative for malignancy, infection, autoimmune or inflammatory conditions, or demyelination. He was diagnosed with radiation-induced myelitis. He was treated with HD-MTX and had a minimal response. Treatment with bevacizumab led to complete resolution of the lesion, and after intensive physical therapy, he regained neurologic function.
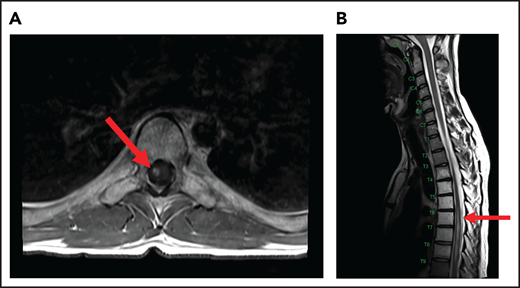
MRI of the cervical and thoracic spine of a patient with radiation-induced myelopathy. (A) Axial T1-weighted image after gadolinium contrast shows enhancement within the spinal cord. (B) Sagittal T2 image shows a signal change throughout the patient’s prior radiation portal from T5 to T9, demarcated by fatty marrow replacement between the T5 and T9 vertebral bodies.
MRI of the cervical and thoracic spine of a patient with radiation-induced myelopathy. (A) Axial T1-weighted image after gadolinium contrast shows enhancement within the spinal cord. (B) Sagittal T2 image shows a signal change throughout the patient’s prior radiation portal from T5 to T9, demarcated by fatty marrow replacement between the T5 and T9 vertebral bodies.
Radiation
Ionizing radiation to the brain or spinal cord can cause acute, subacute, or delayed adverse effects.58 Acute brain toxicity from radiation is typically related to cerebral edema and/or raised intracranial pressure and resolves with corticosteroids. Subacute effects caused by radiation-induced necrosis occur weeks to months after radiation and usually present with headache, lethargy, and focal neurologic deficits. Rarely, radiation myelopathy can result from focal radiation delivered to the mediastinum for HL and NHL, as in case 5.61 Spine MRI typically shows contrast enhancement within the spinal cord, and symptoms may resolve with corticosteroids. Bevacizumab has been used to treat radiation necrosis, as described in our case.62 Delayed neurotoxicity typically presents years after treatment. It can present as radiation-induced vasculopathy resulting in strokes or hemorrhage, or as brain atrophy and leukoencephalopathy. Older age and concurrent use of HD-MTX increase this risk.63
In summary, patients with lymphoma are at high risk of neurologic complications from direct or indirect effects, including treatment-related toxicities. With an increasing number of US Food and Drug Administration–approved treatments for lymphoma, awareness of rare side effects that can lead to significant neurologic morbidity or even mortality is important. Consideration of a broad differential diagnosis for new onset or exacerbation of existing neurologic symptoms is advisable along with early intervention and involvement of appropriate neurological specialists.
Acknowledgment
L.N. is supported by a Leukemia and Lymphoma Society Career Development Program (CDP) grant (2322-19).
Authorship
Contribution: L.N. and T.T.B. wrote the paper.
Conflict-of-interest disclosure: L.N. has consulted for Ono Pharmaceutical, has received clinical trial research support from Merck, Kazia and Astra Zeneca, and delivered Continuing Medical Education (CME) lectures for Oakstone Medical Publishing and Neurology Audio Digest. T.T.B. has consulted for Jiahui Health, Genomicare, and SAB; has delivered CME lectures and provided materials for UpToDate Inc, Oncology Audio Digest, and Oakstone Medical Publishing.
Correspondence: Tracy T. Batchelor, Brigham and Women’s Hospital, Hale Building for Transformative Medicine, 4th Floor, 60 Fenwood Rd, Boston, MA 02113; e-mail: tbatchelor@bwh.harvard.edu.