Key Points
Liso-cel was associated with manageable toxicities and rapid and deep responses in patients with relapsed/refractory CLL/SLL.
With successful manufacturing of liso-cel for patients with CLL/SLL and the encouraging phase 1 results, a phase 2 study is underway.

Abstract
Bruton tyrosine kinase inhibitors (BTKi) and venetoclax are currently used to treat newly diagnosed and relapsed/refractory chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL). However, most patients eventually develop resistance to these therapies, underscoring the need for effective new therapies. We report results of the phase 1 dose-escalation portion of the multicenter, open-label, phase 1/2 TRANSCEND CLL 004 (NCT03331198) study of lisocabtagene maraleucel (liso-cel), an autologous CD19-directed chimeric antigen receptor (CAR) T-cell therapy, in patients with relapsed/refractory CLL/SLL. Patients with standard- or high-risk features treated with ≥3 or ≥2 prior therapies, respectively, including a BTKi, received liso-cel at 1 of 2 dose levels (50 × 106 or 100 × 106 CAR+ T cells). Primary objectives included safety and determining recommended dose; antitumor activity by 2018 International Workshop on CLL guidelines was exploratory. Minimal residual disease (MRD) was assessed in blood and marrow. Twenty-three of 25 enrolled patients received liso-cel and were evaluable for safety. Patients had a median of 4 (range, 2-11) prior therapies (100% had ibrutinib; 65% had venetoclax) and 83% had high-risk features including mutated TP53 and del(17p). Seventy-four percent of patients had cytokine release syndrome (9% grade 3) and 39% had neurological events (22% grade 3/4). Of 22 efficacy-evaluable patients, 82% and 45% achieved overall and complete responses, respectively. Of 20 MRD-evaluable patients, 75% and 65% achieved undetectable MRD in blood and marrow, respectively. Safety and efficacy were similar between dose levels. The phase 2 portion of the study is ongoing at 100 × 106 CAR+ T cells. This trial was registered at clinicaltrials.gov as NCT03331198.
Introduction
Targeted therapies, such as Bruton tyrosine kinase inhibitors (BTKi; eg, ibrutinib, acalabrutinib), phosphatidylinositol 3-kinase inhibitors (PI3Ki; eg, idelalisib, duvelisib), and the B-cell lymphoma 2 inhibitors (eg, venetoclax), alone or combined with CD20 monoclonal antibodies, have improved outcomes for patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL).1-3 In the United States, these targeted therapies have largely replaced chemoimmunotherapy as the preferred treatment of previously untreated and relapsed/refractory CLL/SLL4 and have significant activity in high-risk patients with del(17p), mutated TP53, unmutated immunoglobulin heavy-chain variable (IGHV) gene, and/or complex karyotype.1,5-7 Although a majority of patients respond to targeted therapies, most patients with relapsed/refractory disease eventually develop intolerance or experience progressive disease (PD), especially patients receiving these treatments as later lines of therapy and those with poor-risk disease features.8 Therefore, there is an unmet need for effective new therapies for these patients.
Although chimeric antigen receptor (CAR) T-cell therapy was successfully developed for the treatment of relapsed/refractory non-Hodgkin lymphoma, clinical studies in CLL remain exploratory.9-12 Lisocabtagene maraleucel (liso-cel), an autologous, CD19-directed, defined-composition CAR T-cell product administered at equal target doses of CD8+ and CD4+ CAR+ T cells, demonstrated durable clinical activity and favorable safety in patients with relapsed/refractory large B-cell lymphoma (LBCL).13 We report results from the phase 1 monotherapy dose-escalation portion of the TRANSCEND CLL 004 study investigating liso-cel in patients with relapsed/refractory CLL/SLL.
Methods
Study design and patients
TRANSCEND CLL 004 is an ongoing phase 1/2, open-label, multicenter study evaluating the safety and efficacy of liso-cel in adults with relapsed/refractory CLL/SLL. The phase 1 dose-escalation portion includes cohorts assessing liso-cel monotherapy and liso-cel combined with ibrutinib. The primary objective of the phase 1 monotherapy portion was to assess safety and determine the recommended dose for the subsequent phase 2 expansion cohort. The primary endpoint was the modified toxicity probability interval-2 (mTPI-2) algorithm output of the type, frequency, and severity of adverse events (AE) and laboratory abnormalities.14 Exploratory endpoints included overall response rate (ORR), complete response (CR) rate,15 and cellular kinetics.
Eligible patients were ≥18 years of age and had relapsed/refractory CLL/SLL, prior BTKi treatment failure (ie, stable disease [SD] or PD as best response, progression after previous response, or discontinuation due to BTKi intolerance) or exposure, adequate organ function, and an Eastern Cooperative Oncology Group performance status of 0 to 1. Patients with high-risk relapsed/refractory CLL/SLL (ie, complex cytogenetics [≥3 clonal chromosomal abnormalities], del(17p), mutated TP53, or unmutated IGHV) must have received ≥2 lines of prior therapy. Patients with standard-risk CLL/SLL must have received ≥3 lines of prior therapy. Exclusion criteria included active central nervous system (CNS) leukemia involvement, active infection, prior gene therapy, Richter transformation (RT), allogeneic hematopoietic stem cell transplant within 100 days prior to leukapheresis, or evidence of graft-versus-host disease. Full eligibility criteria are listed in supplemental Methods available on the Blood Web site.
The study was registered with clinicaltrials.gov (NCT03331198) and conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice guidelines, and applicable regulatory requirements. The protocol and its amendments were approved by an Institutional Review Board at each study site. Patients provided written informed consent before study participation.
Study treatment
Patients underwent leukapheresis for liso-cel production (supplemental Methods); no absolute lymphocyte count restrictions were required. Bridging therapy was permitted after leukapheresis, during liso-cel production, and before lymphodepleting chemotherapy (LDC). The washout period varied according to the type of bridging therapy used and was 7 days for most options (range, 3-7 days; supplemental Table 1; available on the Blood website). Baseline assessment, including confirmation of measurable disease, was required before initiating LDC. Following successful manufacturing of liso-cel, patients received 3 days of LDC with fludarabine (30 mg/m2 per day) plus cyclophosphamide (300 mg/m2 per day). Of 4 planned dose levels (DL) of liso-cel (supplemental Methods), patients received 50 × 106 (DL1) or 100 × 106 (DL2) CAR+ T cells as 2 sequential intravenous infusions of CD8+ and CD4+ CAR+ T cells 2-7 days after LDC. Products in which 1 of the CD8 or CD4 cell components did not meet the requirements to be considered liso-cel were defined as nonconforming. Patients who received nonconforming product were not included in the safety or efficacy analysis. Liso-cel retreatment was allowed if patients achieved a CR after liso-cel treatment and later progressed, including progression by RT.
Assessments
Dose-limiting toxicities (DLT) are defined in supplemental Methods. The DLT-evaluable analysis set included all patients who received liso-cel and had either experienced a DLT or were followed for the full 28-day DLT evaluation period. Patients were monitored for toxicities via physical examination, blood work, and Mini-Mental State Examination at specific time points (supplemental Table 2).
High-risk disease was defined as complex karyotype (≥3 chromosomal abnormalities), del(17p), mutated TP53, or unmutated IGHV and was locally assessed. Prolonged cytopenia was defined as grade ≥3 anemia, neutropenia, or thrombocytopenia not resolved at the day 30 study visit; these events were considered resolved when they decreased to grade ≤2. Neurological events (NE) were defined as investigator-identified neurological AEs related to liso-cel. Treatment-emergent adverse events (TEAE) and NEs were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03. Cytokine release syndrome (CRS) was graded according to the Lee 2014 criteria.16 Management of CRS and NEs included early intervention for grade 2 CRS using tocilizumab to prevent progression to higher grade CRS (supplemental Methods). The β2 microglobulin, anemia, lactate dehydrogenase, and last therapy (BALL) prognostic score was calculated post hoc (supplemental Methods).17,18
Response was based on investigator assessment using International Workshop on Chronic Lymphocytic Leukemia criteria15 on day 30 and periodically reassessed until PD, study discontinuation, or death (supplemental Table 2). Response assessments included diagnostic quality computed tomography scans. Minimal residual disease (MRD; sensitivity of 10−4) was assessed in peripheral blood by flow cytometry (Cancer Genetics, Inc./Interpace Diagnostics, Rutherford, NJ) and in marrow by next-generation sequencing (NGS) (Adaptive Biotechnologies, Seattle, WA) in all patients at day 30 and at subsequent time points (supplemental Table 2).
Peripheral blood was sampled to evaluate liso-cel cellular kinetics using validated quantitative polymerase chain reaction to detect the liso-cel transgene in peripheral blood (supplemental Table 2).13,19 Cellular kinetic parameters were estimated from the individual transgene-time profiles using a noncompartmental analysis including area under the curve from 0 to 28 days postinfusion (AUC0‒28d), maximum expansion (Cmax), and time to Cmax (tmax). Liso-cel persistence at each time point was defined as a transgene count greater than or equal to the lower limit of detection.
Patients were followed for 2 years after their last liso-cel dose for safety, efficacy, subsequent anticancer therapies, and survival and were subsequently invited to enroll in a long-term, 15-year follow-up study (NCT03435796).
Statistical analysis
A maximum sample size of 44 patients evaluable for DLTs was permitted; dose escalation or de-escalation was determined using the mTPI-2 statistical design,14 with a 30% target DLT rate and an equivalence interval of 25% to 35%. A DL was considered unsafe, with no additional patients enrolled at that DL, if it had an estimated ≥95% probability of exceeding the target DLT rate of 30% with ≥3 patients treated at that DL. Liso-cel dose escalation could be halted once the recommended dose was selected. Descriptive summaries were provided for demographics, baseline characteristics, patient disposition, safety, antitumor activity, and cellular kinetics. The Kaplan-Meier method was used to assess duration of response (DOR) and progression-free survival (PFS).
Results
Patients
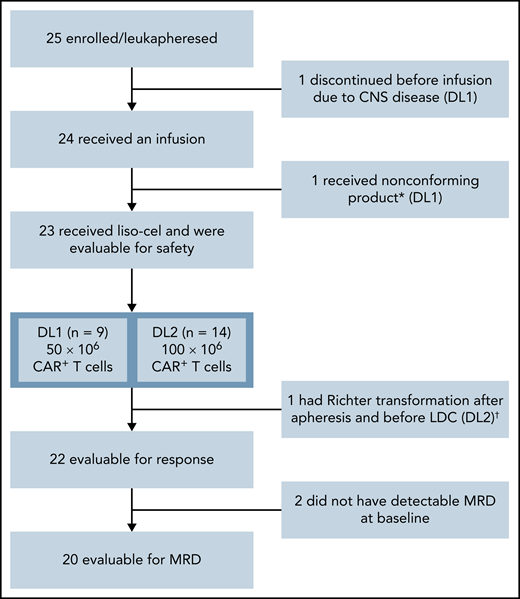
Twenty-five patients were enrolled from January 2018 (first patient received leukapheresis) to January 2019 (last patient treated) at 8 US study sites. One patient discontinued the study prior to liso-cel infusion because of CNS disease. Liso-cel was successfully manufactured for 23/24 patients, and 1 patient received nonconforming product and was excluded from the safety-evaluable population (n = 23) (Figure 1). Of 23 patients who received liso-cel, 9 were treated at DL1 (50 × 106 CAR+ T cells) and 14 at DL2 (100 × 106 CAR+ T cells). One patient at DL1 who initially achieved CR with liso-cel and subsequently progressed was re-treated with liso-cel but did not respond to retreatment.
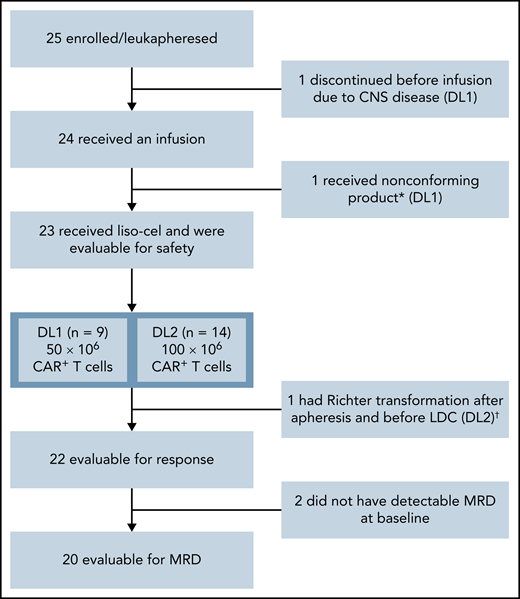
Patient flow diagram. *Liso-cel is composed of an equal target dose of CD8+ and CD4+ CAR+ T cells. While CAR+ T cells could be manufactured for all patients, 1 patient received nonconforming product. †This patient was discovered to have Richter transformation after leukapheresis and prior to receiving liso-cel and was included in the safety population but not the efficacy population, as Richter transformation was an exclusion criterion.
Patient flow diagram. *Liso-cel is composed of an equal target dose of CD8+ and CD4+ CAR+ T cells. While CAR+ T cells could be manufactured for all patients, 1 patient received nonconforming product. †This patient was discovered to have Richter transformation after leukapheresis and prior to receiving liso-cel and was included in the safety population but not the efficacy population, as Richter transformation was an exclusion criterion.
The median age was 66 years, and 11 patients (48%) were male (Table 1). Twenty-one patients (91%) had nodal disease, 8 (35%) had bulky (>5 cm) lymph nodes, and 13 (57%) had disease in the marrow. Six patients had hypogammaglobulinemia at baseline. The median prognostic BALL risk score was 2 (range, 0-3). Nineteen patients (83%) had high-risk genetic features, including mutated TP53, del(17p), complex karyotype, and/or unmutated IGHV. All 8 patients with del(17p) also had mutated TP53. Patients had a median of 4 (range, 2-11) lines of prior therapy. All patients had received prior ibrutinib (91% refractory or relapsed), 20 (87%) had prior chemoimmunotherapy, and 15 (65%) had both prior ibrutinib and venetoclax. Two patients previously treated with ibrutinib were intolerant only and 21 were relapsed/refractory to ibrutinib. Of the 21 patients who were relapsed/refractory to ibrutinib, 4 were also intolerant to prior ibrutinib. Seventeen patients (74%) received bridging therapy during liso-cel manufacturing (supplemental Table 3).
Safety
No DLTs were reported in patients who received DL1. DLTs were reported in 2 patients treated at DL2 (1 patient had grade 4 hypertension, and 1 patient had grade 3 encephalopathy, grade 3 muscle weakness, and grade 4 tumor lysis syndrome). All DLTs completely resolved. Per the mTPI-2 algorithm, the DLT rate of 2 of 14 patients treated at DL2 did not warrant dose de-escalation, and 100 × 106 CAR+ T cells was selected as the recommended dose in consultation with the safety review committee.
Among 23 safety-evaluable patients, the most frequent TEAEs occurring in more than half of patients were anemia (83% [n = 19]), CRS (74% [n = 17]), thrombocytopenia (74% [n = 17]), and neutropenia/neutrophil count decrease (70% [n = 16]; Table 2). The most common grade 3/4 TEAEs were cytopenias, including anemia (74% [n = 17]), thrombocytopenia (70% [n = 16]), neutropenia/neutrophil count decrease (70% [n = 16]), leukopenia (43% [n = 10]), febrile neutropenia (26% [n = 6]), and lymphopenia (26% [n = 6]). The safety profile observed for patients at the 2 DLs was similar (Table 2; supplemental Tables 4-6). Serious TEAEs of any grade were reported in 57% (n = 13) of patients across both DLs (supplemental Table 4).
Nine deaths occurred; 7 patients died of PD. One patient developed grade 3 pneumonia (DL1) 2 months after receipt of liso-cel and died 2 weeks later with grade 5 respiratory failure considered unrelated to liso-cel treatment. Lymphodepleting chemotherapy (both cyclophosphamide and fludarabine) were suspected causes of the pneumonia. The second patient (DL2) with acute kidney injury and pneumonia died >90 days after liso-cel infusion from septic shock considered unrelated to liso-cel treatment. At the last visit for which data were collected, which occurred ∼3 months before death, the patient had grade 1 neutropenia. The neutropenia resolved to normal levels a few days later. No deaths occurred within the first 30 days. Two patients had an ongoing NE at the time of death and 3 had RT.
Seventeen patients (74%) developed any-grade CRS, with a median onset of 3 days (range, 1-10), which resolved in all patients. Two patients (9%; both at DL2 [14%]) developed grade 3 CRS (Table 3). No grade >3 CRS events were reported. CRS symptoms are shown in supplemental Table 7. Nine patients (39%) had any-grade NEs, with a median onset of 4 days (range, 2-21). Four patients (17%; 2 patients at DL2 [14%]) developed grade 3 NEs (muscular weakness, confusional state, encephalopathy, aphasia) (supplemental Table 8). One patient (4%) treated at DL2 had grade 4 NEs (somnolence, encephalopathy). Eight patients (35%) reported >1 NE episode (median [range], 3 [2-11]). Eight patients (35%) experienced both CRS and NEs; CRS generally occurred prior to NEs.
Fifteen patients (65%) received tocilizumab and/or corticosteroids to manage CRS and/or NEs. Six patients (26%) received tocilizumab only, 1 (4%) received corticosteroids only, and 8 (35%) received both therapies. Vasopressors were used in 1 patient (4%). Owing to early intervention in CRS management, 12 of 15 patients with grade 1 or 2 CRS received tocilizumab, with or without corticosteroids, compared with 2 of 2 patients with grade 3 CRS.
Other TEAEs of special interest included hypogammaglobulinemia (22% [n = 5]), grade ≥3 infections (30% [n = 7]), tumor lysis syndrome (17% [n = 4]), and prolonged cytopenias (78% [n = 18]). Thirteen patients required intravenous immunoglobulin repletion. Nine of 12 patients (75%) at 12 months and 3 of 7 (43%) at 24 months had B-cell aplasia (supplemental Table 9). Of 18 patients with prolonged cytopenias, all required transfusions, 11 (61%) required growth factor support, and 13 (72%) resolved to grade ≤2 by day 90.
Median duration of hospital stay for liso-cel administration was 17 days (range, 8-75) (Table 4). After initial discharge, 12 patients (52%) were rehospitalized. One patient was rehospitalized to receive a subsequent anticancer therapy (venetoclax). Eleven patients (48%) were rehospitalized due to AEs, 5 (22%) of whom were rehospitalized for CRS and/or NEs (all grade ≤3). The AEs that resulted in rehospitalization for the 6 remaining patients included pneumonia (n = 2), febrile neutropenia (n = 2), lung infection and fever (n = 1), and fever and febrile neutropenia (n = 1). Two patients rehospitalized for CRS and/or NE also had other AEs for which they were rehospitalized at the same time: decreased fibrinogen and hyponatremia (n = 1) and tumor lysis syndrome (n = 1). The median time to first rehospitalization due to an AE after liso-cel infusion was 28 days (range, 3-241), and the median duration of first rehospitalization due to an AE was 6 days (range, 3-14). Two patients required admission or transfer to the ICU after liso-cel administration for toxicity management. One patient treated at DL1 had grade 4 pneumonia and was admitted to the ICU on day 29 for 14 days. One patient treated at DL2 was transferred to the ICU on day 4 for 9 days for grade 3 CRS and grade 4 tumor lysis syndrome, both of which resolved.
Efficacy
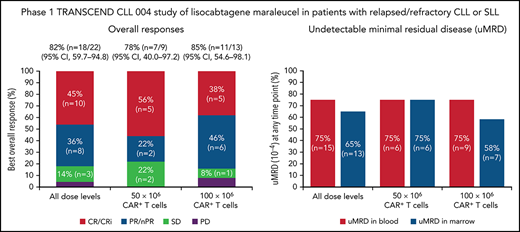
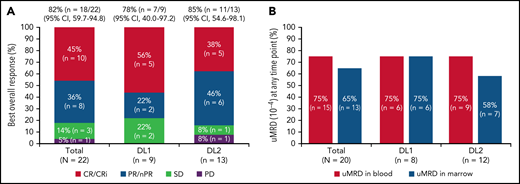
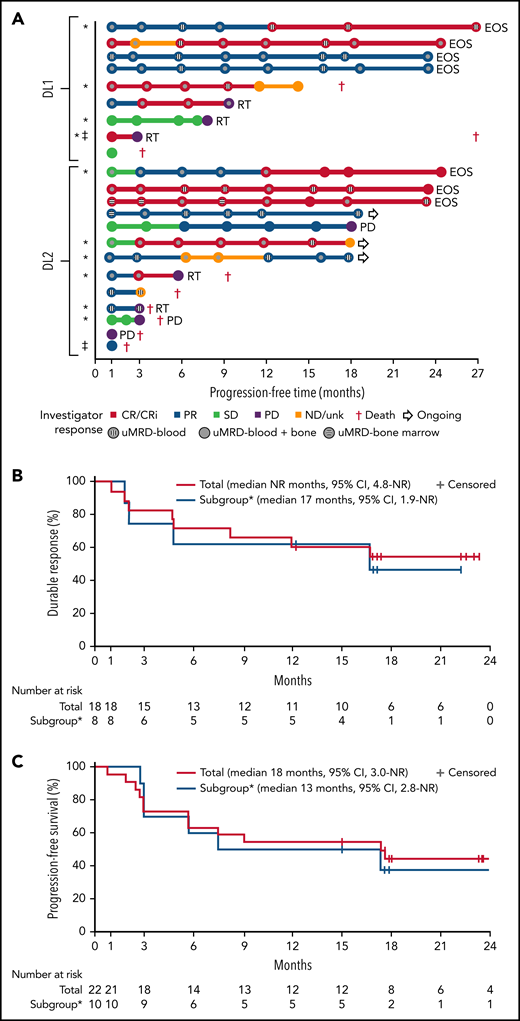
One patient had RT diagnosed after leukapheresis, but before LDC, and was excluded from the efficacy analysis. All 22 efficacy-evaluable patients had measurable disease at baseline. The median follow-up was 24 months (95% confidence interval [CI], 17.9-24.4) (data cutoff, 24 July 2020). The ORR was 82% (95% CI, 59.7-94.8) and the CR/CR with incomplete marrow recovery (CRi) rate was 45% (6 CR and 4 CRi) (Figure 2A). Responses were rapid and durable (Figure 3A), with a median time to response of 1 month (range, 1-6) and median time to CR/CRi of 2 months (range, 1-12). Fifteen patients (68%) achieved a response by day 30 and 6 (27%) experienced deepening of responses over time (4 PRs became CRs [2 at month 3; 2 at month 12], 1 SD became a PR at month 6, and 1 SD improved to CR at month 3). Five of 22 patients (23%) progressed with RT. Three patients who progressed with RT achieved CR before transformation, 1 had SD for >6 months, and 1 had no response. Among 14 patients with a response at 6 months, 12 (86%) maintained their response at 12 months and 2 progressed (with RT). At time of data cutoff, 11 patients had ≥18 months of follow-up, including 7 who completed the study. Of these 11 patients, 10 (45% of all 22 efficacy-evaluable patients) were still in response at 18 months, and 1 patient had progressed. Of the 7 patients who completed the 24-month study, 5 remained in CR and 2 remained in PR. One patient had an unknown response at the last study visit between 12 and 15 months and later died at 18 months. With a median follow-up of 24 months, the median DOR was NR, and the median PFS was 18 months (95% CI, 3.0-NR) (Figure 3B-C).
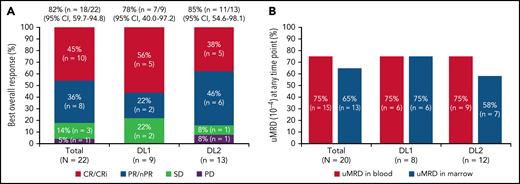
Responses and uMRD. (A) Best overall response. Response-evaluable patients were defined as having had a pretreatment assessment and ≥1 postbaseline assessment. One patient in the safety population was not evaluable for response. (B) uMRD. MRD-evaluable patients were defined as those with detectable MRD at baseline. Two patients in the response-evaluable population were not evaluable for MRD. nPR, nodular partial response; PR, partial response; uMRD, undetectable minimal residual disease.
Responses and uMRD. (A) Best overall response. Response-evaluable patients were defined as having had a pretreatment assessment and ≥1 postbaseline assessment. One patient in the safety population was not evaluable for response. (B) uMRD. MRD-evaluable patients were defined as those with detectable MRD at baseline. Two patients in the response-evaluable population were not evaluable for MRD. nPR, nodular partial response; PR, partial response; uMRD, undetectable minimal residual disease.
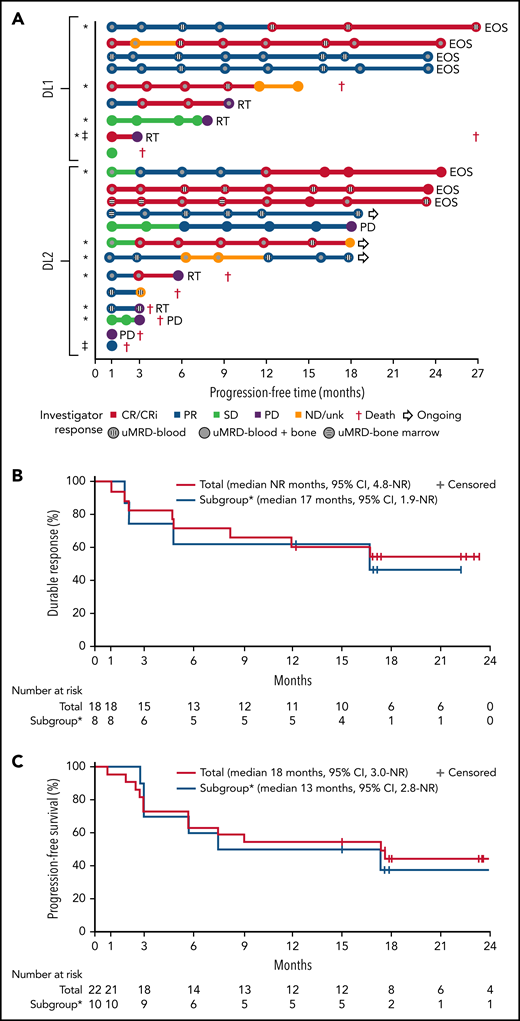
Swim lane plot, duration of response, and progression-free survival. (A) Individual patient response assessments, (B) duration of response, and (C) progression-free survival. *BTKi progression/venetoclax failure subgroup, defined as patient(s) whose disease progressed on BTKi and had failed venetoclax due to progression, intolerance, or failure to respond after ≥3 months of therapy. ‡MRD nonevaluable patients. No patient who achieved uMRD and was tested subsequently had detectable disease at a later time point. Testing for MRD was not required after month 12. EOS, end of study; ND/unk, not done/unknown; NR, not reached; RT, Richter transformation.
Swim lane plot, duration of response, and progression-free survival. (A) Individual patient response assessments, (B) duration of response, and (C) progression-free survival. *BTKi progression/venetoclax failure subgroup, defined as patient(s) whose disease progressed on BTKi and had failed venetoclax due to progression, intolerance, or failure to respond after ≥3 months of therapy. ‡MRD nonevaluable patients. No patient who achieved uMRD and was tested subsequently had detectable disease at a later time point. Testing for MRD was not required after month 12. EOS, end of study; ND/unk, not done/unknown; NR, not reached; RT, Richter transformation.
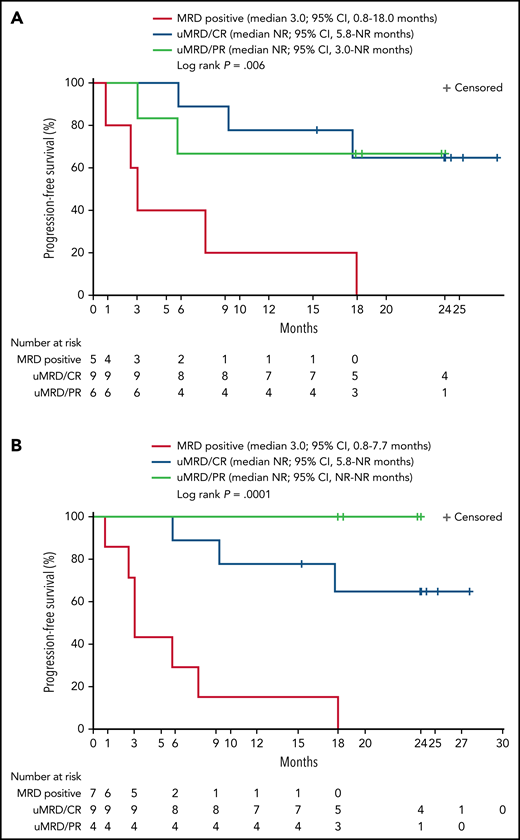
Twenty patients were evaluable for MRD; 2 patients (1 from each DL) did not have detectable disease in peripheral blood or marrow (ie, measurable disease by lymph node only) by MRD testing at baseline and were excluded from the MRD analysis. Overall, 15 patients (75%) achieved uMRD (sensitivity <10−4) in blood, of which 13 (65%) also achieved uMRD in marrow (Figure 2B). Most patients achieved uMRD rapidly, with 13 achieving uMRD in blood and 12 achieving uMRD in marrow by day 30. Of the 13 patients who achieved uMRD in both blood and marrow at any time, 3 have progressed (2 with RT). Patients who achieved uMRD in blood or marrow and had a best response of PR or CR had a better median PFS than those who did not achieve uMRD (NR vs 3 months for uMRD assessed in blood and marrow, respectively; Figure 4A-B).
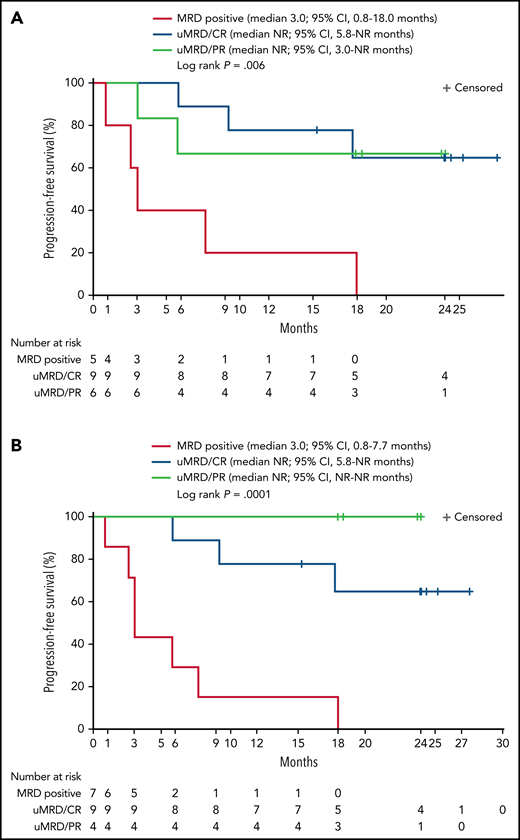
Progression-free survival according to detectable disease status by flow cytometry and NGS. Patients were grouped according to whether MRD was detectable (MRD positive) or undetectable (uMRD; <10−4) in peripheral blood as analyzed by flow cytometry and by response (A) or detectable (MRD positive) or undetectable (uMRD; <10−4) in bone marrow as analyzed by NGS and by response (B). Response was classified as CR or PR.
Progression-free survival according to detectable disease status by flow cytometry and NGS. Patients were grouped according to whether MRD was detectable (MRD positive) or undetectable (uMRD; <10−4) in peripheral blood as analyzed by flow cytometry and by response (A) or detectable (MRD positive) or undetectable (uMRD; <10−4) in bone marrow as analyzed by NGS and by response (B). Response was classified as CR or PR.
All 8 patients with del(17p) also had mutated TP53. Among the 14 patients with mutated TP53, 6 achieved CR/CRi (5 CR and 1 CRi), 4 achieved PR, 3 had SD, and 1 was not evaluable for response because of RT discovered after enrollment but before liso-cel infusion.
No specific risk factors for RT were identified. No baseline factors were found to be significant (age, number or type of prior therapies, sum of the product of perpendicular diameters, risk score), though analyses are limited by small sample size. Of the 5 patients with RT, 4 were in the subgroup of patients whose disease had progressed on BTKi and failed venetoclax. Transformation occurred after an initial response to liso-cel in 3 of these patients. Two patients were at standard risk (4 and 6 prior lines of therapy, respectively) and 3 patients had at least 1 high-risk feature (4, 5, and 7 prior lines of therapy, respectively). Four patients with RT received salvage therapies after progressing on liso-cel. These therapies included pembrolizumab; umbralisib plus ublituximab; everolimus; pomalidomide plus rituximab plus polatuzumab; obinutuzumab; cyclophosphamide plus doxorubicin plus vincristine plus prednisone; and BTKi (unknown).
Cellular kinetics
Among 23 evaluable patients, median Cmax, AUC0‒28d, and tmax were 55 100 copies/µg, 392 000 day*copies/µg, and 14.5 days at DL1 (n = 9), respectively, and 67 400 copies/µg, 643 000 day*copies/µg, and 15 days at DL2 (n = 14), respectively (supplemental Table 10). Higher median peak CAR T-cell expansion and overall CAR T-cell exposure were observed in patients treated at DL2 compared with those at DL1; however, considerable interpatient variability in cellular kinetic parameters was observed at both DLs. Among patients with evaluable samples, CAR+ T cells showed persistence at 12 months in 2 of 4 patients treated at DL1 and 4 of 8 treated at DL2. CAR+ T cells were not detected in any patient at 24 months (n = 7; 4 at DL1 and 3 at DL2) (supplemental Table 11).
Outcomes of patients in the BTKi progression/venetoclax failure subgroup
Further analysis was performed in a subset of 11 patients (48%) whose disease progressed on BTKi and failed venetoclax because of progression, intolerance, or failure to respond after ≥3 months of therapy. Two patients (18%) in this subgroup developed grade 3 CRS and 3 (27%) developed grade 3 or 4 NEs (confusional state, encephalopathy, aphasia, muscular weakness) (supplemental Table 12). Of the 10 patients evaluable for response, best ORR was 80% (95% CI, 44.4-97.5) and CR/CRi rate was 60%; 78% achieved uMRD in blood and 67% in marrow (supplemental Table 13). Five of these patients had a response for ≥6 months (Figure 3A). Four of the 5 patients who progressed with RT were in this subgroup of patients. The median DOR and PFS in this subgroup was 17 (95% CI, 1.9-NR) months and 13 (95% CI, 2.8-NR) months, respectively (Figure 3B-C). Cellular kinetic data are shown in supplemental Table 14.
Discussion
TRANSCEND CLL 004 is the first multicenter trial of CD19-directed CAR T-cell therapy in heavily pretreated patients with relapsed/refractory CLL/SLL. Liso-cel was successfully manufactured for 23 of 24 patients (96%) using the same process as for the manufacturing of liso-cel for patients with LBCL.20,21 Early and deep responses were observed in patients with high-risk disease, most of whom were previously treated with BTKi and B-cell lymphoma 2 inhibitors. Most patients achieved uMRD in blood and/or marrow by the day 30 disease assessment. The median PFS was 18 months (95% CI, 3.0-NR) in the overall population, whereas the median PFS in patients with a best response of PR or CR who achieved uMRD in blood and/or marrow was not reached compared with 3 months in those who remained MRD positive. These findings suggest that achieving uMRD may be important in predicting long-term clinical benefit. However, longer follow-up in a larger number of patients is needed to determine the durability of responses with liso-cel in relapsed/refractory CLL/SLL. Overall, the types of AEs observed post–liso-cel treatment in patients with relapsed/refractory CLL/SLL are comparable to those seen with other CD19-directed CAR T-cell products12 and with liso-cel in patients with LBCL.13 Rates of severe CRS and NEs were 9% and 21%, respectively, with no grade 4 CRS and only 1 grade 4 NE. There were no fatal CRS or NE episodes. Approximately half of patients were readmitted to the hospital for AEs after liso-cel infusion, including 22% for CRS and/or NEs. Given the safety profile of liso-cel, some patients are being treated and managed in the outpatient setting in the phase 2 portion of this trial.
Based on cumulative safety and efficacy data in the 23 patients treated at DL1 and DL2, DL2 (100 × 106 CAR+ T cells) was selected as the recommended dose in consultation with the safety review committee. Additionally, median peak CAR T-cell expansion was higher in patients receiving DL2 than in those receiving DL1. Although the sample size in DL1 and DL2 in this study are too small to show meaningful correlation between CAR T-cell expansion and response, this correlation has been observed in larger phase 1 studies with liso-cel.13
The therapeutic landscape for CLL/SLL has evolved significantly over the past 10 years, with BTKis and venetoclax being used in the first-line setting, alone or in combination with each other and/or CD20 monoclonal antibodies.22-24 Although most patients with CLL/SLL respond to single-agent ibrutinib or venetoclax, with mostly PRs, patients ultimately relapse/progress and require subsequent therapy for disease control. Prognosis is especially poor for patients in whom both BTKis and venetoclax have failed. Treatment options such as PI3Kis (eg, idelalisib and duvelisib) do not offer cures and may cause serious AEs. Although potentially curative, toxicities associated with allogeneic hematopoietic stem cell transplant may contraindicate its use in older patients. In this context and based on the results achieved in relapsed/refractory LBCL, CAR T-cell therapy may provide a potential new treatment option for patients with relapsed/refractory CLL.
Several single-center, single-arm studies investigated other CD19-directed CAR T-cell therapies, utilizing either the CD28 or 4-1BB costimulatory domain, in patients with relapsed/refractory CLL. In these studies, any-grade CRS was reported in 64% to 100% of patients and grade ≥3 CRS in 8% to 24% of patients,10-12 whereas tocilizumab was reported to be administered in 17% to 25% of patients.9,11,12 However, it should be noted that different CRS grading scales (Penn grading scale,11 Memorial Sloan Kettering,25 and Lee 2014 criteria16) were used, which made it difficult for a direct comparison of the incidence of severe CRS across these studies. Grade 3 and higher NEs were reported in 6% to 25% of patients.10-12 Across studies, the ORR ranged from 38% to 74%, and CR rates were low (<30%).9,11 The median PFS reported for evaluable patients was short and ranged from 1 to 8.5 months.9,11,12 However, patients who achieved CR experienced longer median PFS,9,12 suggesting that depth of response may correlate with long-term clinical benefit. In Turtle et al, patients with CR and PR had longer median PFS (9.8 months and NR, respectively) and median OS (NR and NR, respectively) after a median of 12 months of follow-up than nonresponders (PFS, 1.1 months; OS, 11.2 months).12 In Frey et al, patients who achieved a CR had prolonged PFS (40.2 months vs 1 month in patients without CR; P < .0001) and OS (NR vs 31 months in patients without CR; P = .035).9
Historically, CAR T-cell therapy has proven challenging in CLL. Based on preclinical research, several hypotheses are possible. CLL is known to impact the T-cell compartment in affected patients, with a shift in T-cell populations toward terminally differentiated phenotypes, inversion of the normal CD4:CD8 ratio, reduced numbers of naïve T cells, and an increased number of effector memory/effector T cells.26 Additionally, autologous T cells in patients fail to mount a strong antitumor response to CLL tumor cells because of downregulation of genes required for T-cell activation, cytotoxicity, and effector function, as well as immunological synapse formation between CLL tumor cells and cognate T cells.27,28 CAR T-cell dysfunction in patients with CLL could therefore be the result of insufficient activation or inactivation of CAR T cells by the CLL tumor cells.26,29-33 Additionally, a patient’s tumor microenvironment and the phenotype of their infused CAR T cells may impact the expansion and endogenous activity of CAR T cells.34-36
The quality and phenotype of infused CAR T-cell product are known to be related to the initial leukapheresis material and process for creating them.37 The autologous nature of CAR T-cell therapy introduces patient-to-patient variability into the manufacturing process, including significant variance in the absolute lymphocyte count and CD4:CD8 ratio of the incoming material,37 thus creating challenges for achieving CAR T-cell manufacturing consistency across patient populations and disease indications. Liso-cel production involves purifying CD8+ and CD4+ cells separately before activation and transduction.20 Selecting for CD8+ and CD4+ T cells early in the liso-cel manufacturing process reduces the variability of CD3+ T-cell frequencies, relative to leukapheresis, before activation and transduction, with a median T-cell purity of 99.2%. CD3+ T-cell enrichment is further increased throughout the rest of the manufacturing process, which used T-cell–specific activation and expansion.38 Thus, despite potentially substantial heterogeneity in patients with CLL starting material cell composition, including variable CD19+ B-cell frequencies, the liso-cel manufacturing process reduces between-lot variability early in the process, which helps to improve consistency of process performance and final product consistency.26 Of note, all patients in the current study had previously received ibrutinib and many had received prior venetoclax. Both agents have been associated with reducing the number of tumor-supportive T-cell subsets and T cells expressing programmed cell death protein 1 or CTLA-4,39,40 which could have improved the quality of the patients’ leukapheresis material or their tumor microenvironment. Additionally, early clinical data from CAR T-cell and ibrutinib combination studies have shown promising results.41,42
This phase 1 portion of TRANSCEND CLL 004 has some limitations, including the single-arm design, small study sample size, and limited follow-up. The ongoing phase 2 monotherapy expansion cohort, currently enrolling at DL2, will provide additional information on the safety and efficacy of liso-cel in patients with relapsed/refractory CLL/SLL.
Rapid and deep responses were observed after a single dose of liso-cel. Longer follow-up in a larger number of patients is needed to determine the durability of responses and the role of uMRD in sustaining responses. The ability to successfully manufacture liso-cel and the safety and efficacy results from this phase 1 portion of our study in patients with relapsed/refractory CLL/SLL are encouraging. We continue to assess liso-cel in the pivotal phase 2 portion of the study.
Acknowledgments
The authors thank the patients and the study sites for their participation in this study.
This study was funded by Juno Therapeutics, a Bristol-Myers Squibb Company. Karen Ventii, and Nancy Price, CMPP, of The Lockwood Group (Stamford, CT) provided medical writing assistance to the authors during the preparation of this manuscript, funded by Bristol Myers Squibb.
Authorship
Contribution: All authors critically reviewed the draft manuscript, approved the final version to be published, and agreed to be accountable for all aspects of the work; T.S., W.G.W., and J.T. contributed to the study conception or design, data acquisition, and the interpretation of data; H.H.G. and L.G. contributed to the study conception or design, data acquisition, data analysis, and the interpretation of data; L.Y. contributed to the study conception or design, data analysis, and the interpretation of data; K.O. contributed to data analysis and interpretation; and J.D.S., K.A.D., D.M.S., P.A.R., J.A., and T.J.K. contributed to data acquisition.
Conflict-of-interest disclosure: T.S. is an advisor for AstraZeneca, BeiGene, Bristol Myers Squibb, and Kite Pharma outside of the submitted work and has participated in speaker’s bureaus for AstraZeneca, Janssen, and Pharmacyclics outside of the submitted work. J.D.S. reports consulting fees from Abbvie, AstraZeneca, Beigene, Bristol Myers Squibb, TG Therapeutics, and Verastem, and research funding from Adaptive Biotechnologies, Beigene, BostonGene, Genentech/Roche, GlaxoSmithKline, Moderna, and TG Therapeutics. K.A.D. reports salary support from Juno Therapeutics, a Bristol-Myers Squibb Company, to her institution for clinical trial efforts. D.M.S. reports having a consulting or advisory role for Pharmacyclics/Janssen, Karyopharm Therapeutics, BeiGene, and Innate Pharma; honoraria from Genentech; and research funding from Acerta Pharma, Gilead Sciences, Karyopharm Therapeutics, Verastem Oncology, and Juno Therapeutics, a Bristol-Myers Squibb Company. P.A.R. reports speaker’s bureau fees for Bayer and Kite Pharma/Gilead Sciences; honoraria from Bayer and Kite Pharma/Gilead Sciences; membership on an entity’s board of directors or advisory committees for Celgene, a Bristol-Myers Squibb Company, Bayer, Novartis, Karyopharm Therapeutics, Takeda, and Verastem Oncology; and his institution has received research funding from Celgene, a Bristol-Myers Squibb Company, Kite Pharma/Gilead Sciences, MorphoSys, Calibr, and Novartis. J.A. reports grants from Juno Therapeutics, a Bristol-Myers Squibb Company, during the conduct of the study and is a consultant or advisor for Juno Therapeutics, a Bristol-Myers Squibb Company, and Regeneron. T.J.K. reports honoraria from Janssen and Pharmacyclics; membership on an entity’s board of directors or advisory committees for AbbVie, AstraZeneca, Genentech, Janssen, Oncternal Therapeutics, Pharmacyclics, VelosBio, and Verastem Oncology; equity ownership in Oncternal Therapeutics and VelosBio; and his institution has received research funding from AbbVie, Genentech, Oncternal Therapeutics, Pharmacyclics, and VelosBio. H.H.G., L.G., L.Y., K.O., and J.T. are employees of Bristol Myers Squibb and hold stock in Bristol Myers Squibb. W.G.W. reports consultancy from Genzyme, honoraria from Sanofi, and research funding from AbbVie, Acerta Pharma, Cyclacel, Genentech, Gilead Sciences, GlaxoSmithKline/Novartis, Janssen, Juno Therapeutics, a Bristol-Myers Squibb Company, Karyopharm Therapeutics, Kite Pharma, Loxo Oncology, miRagen Therapeutics, Oncternal Therapeutics, Pharmacyclics, Sunesis Pharmaceuticals, and Xencor.
Correspondence: Tanya Siddiqi, Hematology/Hematopoietic Cell Transplantation, City of Hope National Medical Center, 1500 E Duarte Rd, Duarte, CA 91010; e-mail: tsiddiqi@coh.org.
Presented in part at the ASH 2020 Virtual Annual Meeting, December 2020.
Bristol Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.