Abstract
Exposure of blood to a foreign surface in the form of a diagnostic or therapeutic biomaterial device or implanted cells or tissue elicits an immediate, evolutionarily conserved thromboinflammatory response from the host. Primarily designed to protect against invading organisms after an injury, this innate response features instantaneous activation of several blood-borne, highly interactive, well-orchestrated cascades and cellular events that limit bleeding, destroy and eliminate the foreign substance or cells, and promote healing and a return to homeostasis via delicately balanced regenerative processes. In the setting of blood-contacting synthetic or natural biomaterials and implantation of foreign cells or tissues, innate responses are robust, albeit highly context specific. Unfortunately, they tend to be less than adequately regulated by the host’s natural anticoagulant or anti-inflammatory pathways, thereby jeopardizing the functional integrity of the device, as well as the health of the host. Strategies to achieve biocompatibility with a sustained return to homeostasis, particularly while the device remains in situ and functional, continue to elude scientists and clinicians. In this review, some of the complex mechanisms by which biomaterials and cellular transplants provide a “hub” for activation and amplification of coagulation and immunity, thromboinflammation, are discussed, with a view toward the development of innovative means of overcoming the innate challenges.
Introduction
The past few decades have seen steady growth in the use of blood-contacting biomedical devices and cellular therapies to improve patient care and quality of life. They may be used for minutes to hours to years, and include, among others, catheters, stents, grafts, extracorporeal devices, heart valves, drug delivery systems, scaffolds for tissue encapsulation, modified cells and transplants. The numbers are high: >5 million central venous catheters are inserted each year in the United States for drug and fluid delivery, intravascular monitoring, hemodialysis, plasmapheresis, and intracardiac pacing. With advances in biomaterials and the growing demands of an aging population, the implantable medical devices market is expected to exceed $160 billion by 2026.1 Not only will the use of blood-contacting biomaterials rise, but similar growth is expected in therapies using stem/progenitor or differentiated cells to restore function for disorders such as diabetes, cancer, liver failure, and organ ischemia.2–4
What has not changed is the challenge that is met when the host encounters a foreign surface, and innate mechanisms go into overdrive to attempt to discard and/or destroy it. This process results in a thromboinflammatory response that jeopardizes the integrity of the therapeutic or diagnostic device or implanted cell, tissue or organ, often with local or systemic harm to the host. Research efforts have escalated to overcome the consequences of this response, to develop preventative strategies and create biocompatible devices and cells that seamlessly interact with the host. Nonetheless, the challenges persist. For example, central venous catheter–related thrombosis still complicates 1% to 5% of all insertions.5 Ventricular assist devices are confounded by embolic stroke, pump thrombosis, and bleeding.6 Inflammation secondary to hemodialysis remains a risk factor for myocardial ischemia and atheroclerosis.7 The impact of progress in this field would be substantial.
Herein, we discuss key elements involved in the thromboinflammatory response when blood contacts a foreign surface, how they are integrated, and how they participate. Although a paradigm has evolved to describe the sequential overlapping steps, there remain many unknowns. Notably, there is no “one-size-fits-all,” as responses vary, based on multiple factors, including the source, size, and type of biomaterial; surface structure and composition; duration of exposure; the vascular bed or tissue involved; and the age, sex, health, and genetics of the host. Thus, relative contributions of the players and pathways remain incompletely understood, with consequent constraints on developing solutions. Nonetheless, this paradigm yields approaches toward the ultimate design of novel personalized means of attaining biocompatible, functional biomaterials.
A paradigm: thromboinflammatory reactions on biomaterials and transplanted cells
A pictorial representation of the complexity and multiple players involved when a biomaterial comes in contact with blood is shown in Figure 1. The mechanisms of this thromboinflammatory response are described below.
![The thromboinflammatory response of blood after exposure to a biomaterial surface. Although seemingly complex, this is a simplified representation of the multiple cellular and molecular interactions that occur when blood meets a biomaterial surface. Immediately upon contacting a biomaterial surface, plasma proteins (eg, fibrinogen, VWF, immunoglobulin [Ig], and complement proteins [C3, C3(H2O), C3b], FXII, and HK) are adsorbed. Complement is activated via all 3 pathways (AP, CP, and LP) as a result of deposition of C3 (C3, C3(H2O), C3b), complement-fixing Ig (recognized by C1q), and binding of MASPs. The activation of the plasma contact system, its key components being HK, PK, and FXII/FXIIa, results in generation of thrombin (IIa) via the intrinsic pathway, as well as PKa, both of which amplify the thromboinflammatory response via several pathways. Platelets rapidly adhere to adsorbed fibrinogen, augmented under high shear rate, by VWF. These are activated by the intrinsic pathway-generated thrombin and C3a and C5a, that also exert proinflammatory and prothrombotic effects on all immune cells via their respective cell-surface receptors (Figure 3). Cytokines and chemokines are released from the platelets and recruit inflammatory leukocytes, which themselves then release more cytokines and chemokines. C5a and C5b-9 (MAC) activate TF on the monocytes/macrophages which were recruited by chemokines that are released by activated platelets and neutrophils, the latter of which may undergo NETosis, thereby further promoting local and systemic inflammation, thrombin generation, and clot formation. Thrombin, generated via the intrinsic and extrinsic (TF-dependent) pathways, also triggers inflammatory responses via PAR signaling on all immune cells. On the right side, adjacent host vascular endothelium with underlying smooth muscle cells, is shown to highlight how its integrity may be jeopardized during the innate thromboinflammatory response.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/13/10.1182_blood.2020007209/5/m_bloodbld2020007209cf1.png?Expires=1767773504&Signature=Y26jl6fxwCKgXxccEB1mxFrh~a~QXn6tOjk3jV0vbUsZ9tWw4kvqkRGRv3u7UkVF9qX2Dg1uK1IsdTlpeKF-iMIzjX8ERCGNnS85VG~zNxbvUT5NLHLV3Mi9Pt5c52-BxuN9r4KVmVlOVQ-zz6W9KLeAWbkmPzzTbpKPBPzlH-mZda9YJQYDy0-rQyVZY~9WJBM88bQTBPARTnSj7r2Kl9aq1OejBfvW09czMKjB56689kTUj3CYa42ox-u1POFyzBXVyphLFVLDD0PO8RTZyOFs0Ddrv3DUPuECLMNpnzCysik7eBbhX-sPdPmhqJs7b2ncVQ4bMr-9i8RTyqFRcQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The thromboinflammatory response of blood after exposure to a biomaterial surface. Although seemingly complex, this is a simplified representation of the multiple cellular and molecular interactions that occur when blood meets a biomaterial surface. Immediately upon contacting a biomaterial surface, plasma proteins (eg, fibrinogen, VWF, immunoglobulin [Ig], and complement proteins [C3, C3(H2O), C3b], FXII, and HK) are adsorbed. Complement is activated via all 3 pathways (AP, CP, and LP) as a result of deposition of C3 (C3, C3(H2O), C3b), complement-fixing Ig (recognized by C1q), and binding of MASPs. The activation of the plasma contact system, its key components being HK, PK, and FXII/FXIIa, results in generation of thrombin (IIa) via the intrinsic pathway, as well as PKa, both of which amplify the thromboinflammatory response via several pathways. Platelets rapidly adhere to adsorbed fibrinogen, augmented under high shear rate, by VWF. These are activated by the intrinsic pathway-generated thrombin and C3a and C5a, that also exert proinflammatory and prothrombotic effects on all immune cells via their respective cell-surface receptors (Figure 3). Cytokines and chemokines are released from the platelets and recruit inflammatory leukocytes, which themselves then release more cytokines and chemokines. C5a and C5b-9 (MAC) activate TF on the monocytes/macrophages which were recruited by chemokines that are released by activated platelets and neutrophils, the latter of which may undergo NETosis, thereby further promoting local and systemic inflammation, thrombin generation, and clot formation. Thrombin, generated via the intrinsic and extrinsic (TF-dependent) pathways, also triggers inflammatory responses via PAR signaling on all immune cells. On the right side, adjacent host vascular endothelium with underlying smooth muscle cells, is shown to highlight how its integrity may be jeopardized during the innate thromboinflammatory response.
The thromboinflammatory response of blood after exposure to a biomaterial surface. Although seemingly complex, this is a simplified representation of the multiple cellular and molecular interactions that occur when blood meets a biomaterial surface. Immediately upon contacting a biomaterial surface, plasma proteins (eg, fibrinogen, VWF, immunoglobulin [Ig], and complement proteins [C3, C3(H2O), C3b], FXII, and HK) are adsorbed. Complement is activated via all 3 pathways (AP, CP, and LP) as a result of deposition of C3 (C3, C3(H2O), C3b), complement-fixing Ig (recognized by C1q), and binding of MASPs. The activation of the plasma contact system, its key components being HK, PK, and FXII/FXIIa, results in generation of thrombin (IIa) via the intrinsic pathway, as well as PKa, both of which amplify the thromboinflammatory response via several pathways. Platelets rapidly adhere to adsorbed fibrinogen, augmented under high shear rate, by VWF. These are activated by the intrinsic pathway-generated thrombin and C3a and C5a, that also exert proinflammatory and prothrombotic effects on all immune cells via their respective cell-surface receptors (Figure 3). Cytokines and chemokines are released from the platelets and recruit inflammatory leukocytes, which themselves then release more cytokines and chemokines. C5a and C5b-9 (MAC) activate TF on the monocytes/macrophages which were recruited by chemokines that are released by activated platelets and neutrophils, the latter of which may undergo NETosis, thereby further promoting local and systemic inflammation, thrombin generation, and clot formation. Thrombin, generated via the intrinsic and extrinsic (TF-dependent) pathways, also triggers inflammatory responses via PAR signaling on all immune cells. On the right side, adjacent host vascular endothelium with underlying smooth muscle cells, is shown to highlight how its integrity may be jeopardized during the innate thromboinflammatory response.
Protein adsorption: laying the foundation
Immediately upon exposure of a biomaterial to blood, a diverse repertoire of plasma proteins adsorbs to the surface within seconds. This initiating event is the key that sets in motion the development of a dynamically changing, progressively thromboinflammatory microenvironment that is mediated by activation of coagulation and innate immunity, with recruitment of platelets and inflammatory leukocytes. These proteins include fibrinogen, high-molecular-weight kininogen (HK), coagulation factor XI (FXI) and FXII/XIIa, plasma prekallikrein (PK), immunoglobulins (IgG, IgM), complement proteins, albumin, vitronectin, fibronectin, apolipoproteins, and von Willebrand factor (VWF).8,9 The adsorbed plasma proteins, which may also be conformationally altered, form the layers of the “protein corona,” the composition of which depends on specific properties of the surface and may vary over time with adsorption and desorption.10,11 Corona protein concentrations rarely correlate with plasma concentrations and may be >1000-fold higher.12 Moreover, proteomics analyses have revealed a multitude of surface-bound proteins, most likely derived from the surrounding, damaged, leaking tissues.13 Most adsorbed proteins have lower affinity to hydrophilic surfaces because of strong binding to water molecules that are not readily replaced by protein molecules. Thus, hydrophobic biomaterial surfaces tend to adsorb more proteins. Other protein-adsorbing features include rougher and more rigid biomaterials and charge.14
Given the diverse nature and abundance of the adsorbed, often structurally altered, proteins, it is not surprising that the protein corona is a nidus for triggering activation of several cellular events and blood-based proteolytic cascades. Because most biomaterial surfaces lack natural antithrombotic/antiinflammatory properties, the cascades are destined to run unregulated.
Complement: an immediate innate response
The time-dependent increase in the thickness of the protein corona is attributable partly to accumulation of complement factors and binding to conformationally altered adsorbed proteins,11 but also directly to the biomaterial. With links to coagulation,15 complement activation plays a central role in the host’s thromboinflammatory response.16
The complement system comprises >40 soluble and membrane-bound proteins. Its activation serves to rapidly recognize and discard foreign substances or damaged cells, recruit inflammatory cells, promote adaptive immunity, and initiate healing (reviewed by Morgan17 and Ricklin et al18) (Figure 2). In brief, complement is activated via the classic (CP), lectin (LP), or alternative (AP) pathways, each initiated upon exposure to molecular patterns or functional groups displayed on the foreign surface with consequent thromboinflammation when blood is exposed to biomaterials (Figure 1). The CP is triggered by recognition of immune complexes and C-reactive protein by the C1q component of a circulating C1 complex, whereupon the proenzymes C1r and C1s become activated. The LP is initiated by carbohydrates, recognized by complexes of mannose-binding lectin (MBL), ficolins, or collectins and MBL-associated serine proteases (MASPs).19 The AP is constitutively active via a “tickover” mechanism with spontaneous formation of C3(H2O). All pathways converge to form a C3 convertase that cleaves C3 to generate a thiol-reactive C3b and the anaphylatoxin C3a. Unless dampened by regulators, C5 is proteolyzed to C5a, a potent anaphylatoxin, and to C5b, which initiates assembly of the C5b-9 membrane attack complex (MAC).19 Healthy host cells are protected against complement-mediated damage by membrane-anchored (eg, CD55, CD59, CD46, and thrombomodulin) and soluble (eg, C4bBP, C1-esterase inhibitor [C1-INH], FI, FH, vitronectin, and clusterin) negative regulators.17,18 Nonbiologic biomaterials or altered transplanted cells lack intrinsic protection against complement activation.

Biomaterial-triggered complement activation pathways. Complement activation occurs via the CP, LP, or AP. All pathways converge to form C3 convertases and then C5 convertases, with release of the potent anaphylatoxins C3a and C5a. Generation of C5b triggers assembly of the C5b-9 MAC which induces lysis, damage, or cell activation. Regulation is achieved at multiple levels by soluble and membrane-associated factors (C1-INH, FH, FI, CD55, CD46, polyphosphate, CD59, clusterin, vitronectin, and thrombomodulin). C1-INH, C1 esterase inhibitor; CRP, C-reactive protein; TAFIa, activated thrombin activatable fibrinolysis inhibitor; TM, thrombomodulin. Readers are referred to 2 reviews for full descriptions of the pathways.16,17
Biomaterial-triggered complement activation pathways. Complement activation occurs via the CP, LP, or AP. All pathways converge to form C3 convertases and then C5 convertases, with release of the potent anaphylatoxins C3a and C5a. Generation of C5b triggers assembly of the C5b-9 MAC which induces lysis, damage, or cell activation. Regulation is achieved at multiple levels by soluble and membrane-associated factors (C1-INH, FH, FI, CD55, CD46, polyphosphate, CD59, clusterin, vitronectin, and thrombomodulin). C1-INH, C1 esterase inhibitor; CRP, C-reactive protein; TAFIa, activated thrombin activatable fibrinolysis inhibitor; TM, thrombomodulin. Readers are referred to 2 reviews for full descriptions of the pathways.16,17
Triggers for complement activation vary with, for example, surface, biorheology, and host factors.20 C3(H2O) and reactive C3b bind to exposed surface nucleophiles (−OH or −NH2), triggering AP activation. Adsorbed complement-fixing IgG and IgM are recognized by C1q, triggering the CP. Polyethylene glycol–grafted (PEGylated) surfaces can generate epitopes for recognition by naturally occurring IgG and also induce activation via the AP and LP.21 The CP can also be initiated on misfolded proteins or negatively charged surfaces via the contact system, whereby FXII is activated to FXIIa, which can activate C1r and the CP.22 Surface-projected polymers (eg, PEGs, poloxamers, and poloxamines), hydration-altered biomaterial surfaces, and modified adsorbed proteins may be recognized by MBL, collectins, and ficolins, inducing LP activation.23
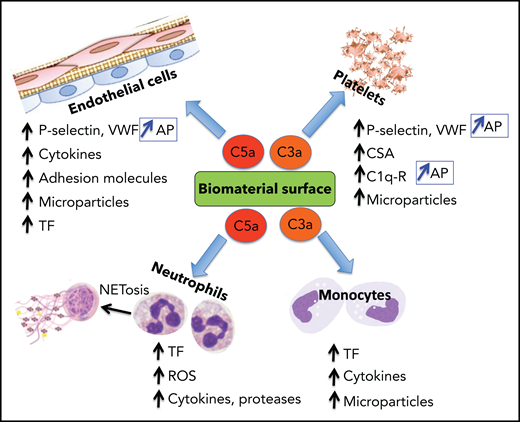
Complement system activation products exert mostly proinflammatory and prothrombotic effects with adverse consequences on biomaterial surfaces. C3a and C5a recruit and activate platelets, polymorphonuclear neutrophils (PMNs), and monocytes (Figure 3). By binding to their cognate receptors, C3a/C5a stimulate leukocyte release of ROS, proteases, proinflammatory cytokines, and tissue factor (TF) expression on leukocytes and in microparticles (MPs), promoting thrombin generation, platelet activation, and clot formation. C3a and C5a also initiate NETosis which drives coagulation, inflammation, and further complement activation. C3a and C5a also provoke secretion of VWF and P-selectin from platelets,24 triggering and sustaining platelet adhesion and aggregation and leukocyte recruitment, the latter via interaction of P-selectin with its receptor PSGL-1.25 P-selectin is also a receptor for C3b, and thus its expression amplifies AP activation.26 Although FH may dampen these effects by enhancing ADAMTS13-mediated proteolysis of ultralarge VWF (ULVWF) multimers,27 reduced ADAMTS13 activity at that site may limit this effect.28
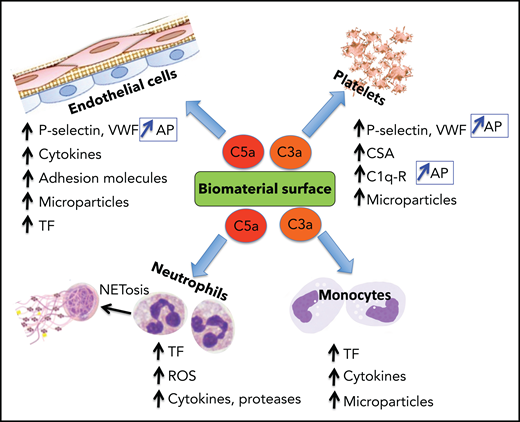
Biomaterial-induced complement activation byproducts C3a and C5a are major mediators of the thromboinflammatory response. The anaphylatoxins C3a and C5a, released during complement activation, induce a wide range of proinflammatory and procoagulant effects, mediated primarily by binding to their cognate receptors. Representative biological effects on platelets, endothelial cells, neutrophils, and monocytes are shown. P-selectin is a receptor for C3b and, when induced by C3a or C5a on platelets and endothelial cells, complement activation is reinforced by assembly of the AP. C1q-r, receptor for C1q; CSA, chondroitin sulfate A; ROS, reactive oxygen species.
Biomaterial-induced complement activation byproducts C3a and C5a are major mediators of the thromboinflammatory response. The anaphylatoxins C3a and C5a, released during complement activation, induce a wide range of proinflammatory and procoagulant effects, mediated primarily by binding to their cognate receptors. Representative biological effects on platelets, endothelial cells, neutrophils, and monocytes are shown. P-selectin is a receptor for C3b and, when induced by C3a or C5a on platelets and endothelial cells, complement activation is reinforced by assembly of the AP. C1q-r, receptor for C1q; CSA, chondroitin sulfate A; ROS, reactive oxygen species.
Terminal pathway C5b-7/9 also promotes coagulation and inflammation, triggering release of chemokines (eg, interleukin-1α [IL1α]), TF activation, and platelet release of VWF, P-selectin, cytokines, and MPs.29 MASPs, which can bind directly to some biomaterial surfaces by unknown mechanisms, directly promote coagulation. MASP-1 and -2 activate prothrombin, whereas MASP-1 activates TAFI, cleaves fibrinogen to fibrin, activates FXIII, and generates bradykinin (BK) from HK. MASPs also complex with ficolins on platelets to activate platelets and complement.30
Overall, excess, unchecked complement activation emanating from the protein corona of blood-exposed biomaterial surfaces triggers a spiral of events, promoting thromboinflammation.
Contact system activation
With protein adsorption occurring at the blood-biomaterial interface, the contact activation and kallikrein-kinin systems, collectively known as the plasma-contact system, trigger coagulation, innate immunity, and inflammation (Figure 4).9 Within seconds of surface exposure, plasma-derived HK and FXII are detectable, often replacing some adsorbed fibrinogen.31 With surface contact, FXII autoactivates, generating FXIIa. FXII is also activated by contact with adsorbed modified fibrinogen, RNA,32 DNA, neutrophil extracellular traps (NETs),33 negatively charged polysaccharides, polyphosphate,34 and activated platelets.35 FXIIa cleaves PK complexed with HK to generate plasma kallikrein (PKa), which feedback generates more FXIIa and PKa. PKa also cleaves C3, C5, and FB, thus activating complement,36–39 and proteolyzes HK to form proinflammatory BK.40 In addition to activating the CP via C1r proteolysis, FXIIa also activates FXI, leading to thrombin generation via the intrinsic pathway. In this context, the intrinsic pathway is important, as the generated thrombin activates platelets, promoting granule secretion of bioactive substances and yielding a surface for prothrombinase assembly and clot formation.41 Thrombin further contributes to thromboinflammation by activating protease-activated receptors (PARs) on recruited leukocytes, which results in phosphorylation of the intracellular kinases42 that induce release of proinflammatory cytokines and chemokines (eg, MCP-1, IL6, IL8, and IL1α). These kinases further recruit platelets and leukocytes, which adhere to the biomaterial surface and promote more thrombin-triggered PAR activation. Finally, thrombin can directly activate complement by cleaving C3, C5,43,44 and pro-IL1α, the latter of which drives inflammation and coagulation by activating monocyte TF (Figure 3).45
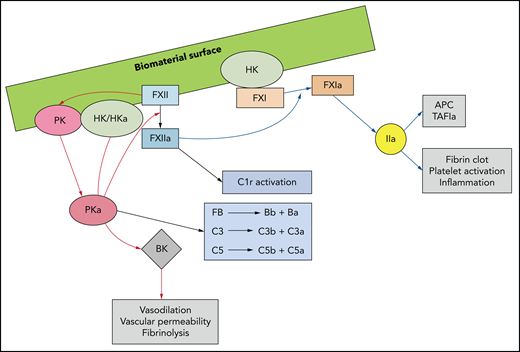
Biomaterial surface–triggered activation of the plasma contact system yields a thromboinflammatory response. Contact of FXII with a biomaterial surface, adsorbed proteins (eg, fibrinogen), DNA, RNA, NETS, polyphosphate, or activated cells, induces a conformational change to generate small amounts of FXIIa. FXIIa complexed with HK activates PK to PKa, FXI to FXIa, and C1r to its active form. PKa cleaves or activates FXII and HK, generating more FXIIa and BK, the latter of which has proinflammatory properties. PKa also directly activates FB, thus promoting the alternative complement pathway and complement factors C3 and C5. FXIa leads to thrombin generation via the intrinsic pathway of coagulation, resulting in activation of adherent platelets. C1-inhibitor (C1-INH) neutralizes PKa, FXIIa, FXIa, and C1r.
Biomaterial surface–triggered activation of the plasma contact system yields a thromboinflammatory response. Contact of FXII with a biomaterial surface, adsorbed proteins (eg, fibrinogen), DNA, RNA, NETS, polyphosphate, or activated cells, induces a conformational change to generate small amounts of FXIIa. FXIIa complexed with HK activates PK to PKa, FXI to FXIa, and C1r to its active form. PKa cleaves or activates FXII and HK, generating more FXIIa and BK, the latter of which has proinflammatory properties. PKa also directly activates FB, thus promoting the alternative complement pathway and complement factors C3 and C5. FXIa leads to thrombin generation via the intrinsic pathway of coagulation, resulting in activation of adherent platelets. C1-inhibitor (C1-INH) neutralizes PKa, FXIIa, FXIa, and C1r.
VWF and platelets
Platelet adhesion to biomaterial surfaces occurs with or immediately after protein adsorption and is the key initial cellular event in the thromboinflammatory response. Mechanisms by which platelets adhere to damaged subendothelium in the circulation are well established and provide insights into their interactions with biomaterial surfaces.46,47 In brief, at low venous shear rates, platelets interact with extracellular matrix collagen, fibronectin, and laminin via GPVI and β1- and β3-integrins. At high arterial shear rates, reversible adhesion is triggered as platelet GPIb-IX-V interacts with unfolded VWF. Stable adhesion requires engagement of collagen receptors (α2β1-integrin) and a fibronectin receptor (α5β1-integrin). Release of platelet agonists (adenosine 5′-diphosphate [ADP], TXA2, and thrombin) triggers inside-out signaling and activation of αIIbβ3-integrin (GPIIb/IIIa), for which there are multiple high-affinity binding sites on fibrinogen and VWF, enabling platelet-platelet interaction and aggregation.46
Normally, resting platelets express GPIIb/IIIa in a closed conformation, with low affinity for its ligands; thus, they do not bind to VWF, fibrinogen, or fibronectin. These platelets, however, can bind to conformationally altered surface-adsorbed fibrinogen, which may occur without VWF.48 Orientation, density, and heterogeneity of adsorbed fibrinogen also influence platelet adhesion. Endothelium-derived factors that normally inhibit adhesion (eg, nitric oxide [NO] and prostacyclin), are absent or present at low levels at the biomaterial surface. Thus, low-affinity platelet adhesion, even with the closed conformation of GPIIb/IIIa, may be sufficient for binding, and other non–Arg-Gly-Asp (RGD) platelet receptors that bind to adsorbed fibrinogen may participate.49
With the low shear flow of blood through polyethylene tubing, low amounts of adsorbed fibrinogen are sufficient to support platelet adhesion, mediated by binding sites for GPIIb/IIIa; adsorbed VWF alone causes low platelet adhesion.49 At high shear rates, VWF, coadsorbed with fibrinogen, greatly enhances platelet adhesion.49 It is believed that adsorbed VWF under shear becomes distorted, facilitating VWF-GPIb/IX interactions which tethers or slows flowing platelets. This effect results in GPIIb/IIIa activation and binding to RGD sequences in VWF and/or adsorption of fibrinogen. Thus, VWF augments platelet adhesion to fibrinogen under high shear.49 These processes are relevant in different biomaterial settings; that is, hemodialysis membranes or arterial grafts are exposed to high shear rates, whereas implant surfaces most often are exposed to low shear rates.
In addition to supporting coagulation propagation,41 platelets are a rich source of cytokines, chemokines, growth factors, and coagulation factors. As thrombin is generated by contact pathway activation, it activates PARs on platelets, triggering mobilization of P-selectin and CD40 ligand; granule release of FV, fibrinogen, FXIII, and polyphosphate; and induction of membrane changes for prothrombinase assembly and thrombin generation. Thrombin-induced platelet release of ADP and TXA2 activates more platelets (Figure 1). Activation of GPIIb/IIIa enables platelets to bind to fibrinogen and VWF, supporting further platelet adhesion, aggregation, and signaling.50 As inflammatory cells enter the mix, TF expressed by monocytes, PMNs and NETs, results in substantial increases in thrombin and fibrin clot formation.
C1qR on activated platelets is recognized by C1q, and, with α-granule–released chondroitin sulfate A, complement activation via the CP is initiated. C1q:C1qR also induces P-selectin release, fueling leukocyte recruitment and AP complement activation.25,26 C3, C4, and C9 bind to activated platelets,51 providing substrates for continued complement activation and proinflammatory and procoagulant effects at the biomaterial surface, with recruitment of more leukocytes and platelets. Complement activation may be dampened via platelet release of the negative regulators C1-INH, FH, CD55, CD59, CD46, clusterin,52 and polyphosphate, although their relevance in blood-biomaterial compatibility is unknown.
Neutrophils
PMNs arrive within minutes to hours at the biomaterial-blood interface, attracted by C3a and C5a and platelet-released chemokines and cytokines (eg, CXCl2, PDGF, and transforming growth factor-β).53 These short-lived effector cells arrive in “swarms,” formed in response to the PMN-released leukotriene LBT4.54 The default action of PMNs is to phagocytose a foreign body. However, unless the target is small (≲350 nm), PMNs become activated via interactions of β2 integrins (LFA-1, Mac-1, and p150/95) with the ligand peptides RGD and Pro-His-Ser-Arg-Asn exposed by adsorbed fibrinogen.55 They also bind to damaged cells or residues of dead cells (damage-associated molecular patterns [DAMPs]) via PMN toll-like receptors (TLRs), the engagement of which triggers NFκB activation and MAPK pathways, releasing proinflammatory factors (IL1β, tumor necrosis factor-α, IL6, IL8, and interferon-γ). These recruit monocytes, inducing their differentiation into proinflammatory M1 macrophages which can express TF, and release factors that recruit and activate more PMNs and other immune cells. This bidirectional, PMN-monocyte cross talk escalates the innate thromboinflammatory response.
In response to many triggers, including foreign surfaces, activated platelets, DAMPs, hypoxia, ROS, and cytokines, PMNs undergo NETosis.56 NETs are secreted, weblike structures, comprising DNA, histones, proteolytic enzymes (eg, neutrophil elastase, myeloperoxidase, and cathepsin G), complement factors, other proteins (eg, lactoferrin), and ions. Substantial evidence implicates NETs in biomaterial-related thromboinflammation, including their presence in thrombosed stents and during hemodialysis.57–59 NETs promote coagulation at the biomaterial surface via multiple mechanisms. They colocalize with fibrin and VWF, providing a scaffold for thrombi that are resistant to fibrinolysis.60 NET contents reduce the capacity of ADAMTS13 to cleave ULVWF multimers.28 They induce activation of platelets and leukocytes, which in turn trigger further NETosis61 and release of prothrombotic polyphosphates.62 Histones interfere with the natural anticoagulants antithrombin (AT), TFPI, and activated protein C.63 DNA from NETs also blocks tPA-mediated plasmin generation and provides a surface for contact pathway activation, augmenting thrombin generation and complement activation.
After 1 to 2 days at the biomaterial surface, PMNs can undergo apoptosis and respond to proinflammatory chemokines and cytokines by releasing molecules that limit further PMN recruitment.64 Those molecules signal M1 macrophages, resulting in efferocytosis of the apoptotic bodies, and polarize to the anti-inflammatory M2 phenotype.65 This important step enables the thromboinflammatory response to the surface to resolve and switch to a more regenerative process for functional integration of the biomedical device or implant.53 If the lifespan of PMNs is prolonged, chronic thromboinflammation ensues. Thus, timely induction of PMN apoptosis and efferocytosis may promote compatibility.
Monocyte/macrophage recruitment
C3a/C5a and chemokines (MIP-1, IFN-γ) released by activated PMNs recruit and activate monocytes (Figures 1, 3, and 4). They, in turn, secrete proinflammatory cytokines,66 and, via β1- and β2-integrins (VLA-4, Mac-1), bind to ligands expressed by adsorbed fibrinogen and fibronectin. They may also adhere directly to biomaterial surfaces.67 With cell surface engagement and in response to PMN-released ROS and HMGB1,68 platelet-secreted cytokines, C5a, and C5b-8/9 complexes, and M1-macrophages upregulate procoagulant TF69 and release TF-laden microvesicles.70,71 The contribution of TF to the immediate consequences of blood-contacting biomaterials is not well understood. However, with sustained thromboinflammation, TF most likely enhances thrombin generation, platelet activation, clot formation, and inflammation. TF-FVIIa triggers PAR signaling, inducing cytokine and chemokine release72 and further upregulation or activation of monocyte TF. This process may lead to biomaterial degradation, device failure, and tissue destruction.
Thromboinflammation on transplanted cells
Although triggers differ, cellular therapies induce similar innate responses that jeopardize survival of the implanted cells and the health of the host. For example, in hepatocyte transplantation, an alternative to liver transplantation,4 an instant blood-mediated inflammatory reaction causes ∼70% hepatocyte loss. In this case, TF that is surface exposed and released by transplanted donor cells is considered the major initiator of the adverse response, activating coagulation, with AP complement activation, platelet adhesion and activation, and inflammatory leukocyte infiltration. Local thrombin generation ensues with clot formation, phagocytic clearance of the transplanted cells, and diminished function of the remaining hepatocytes, with systemic inflammation and coagulation activation.4,73 Similar responses occur with infused mesenchymal stem and CAR-T cells74 and with microencapsulated, implanted pancreatic islet cells.75,76
Preventing thromboinflammation at biomaterial surfaces, now and in the future
Systemic approaches
Systemic therapies to reduce biomaterial-triggered thrombosis are widely used and have primarily been aimed at reducing platelet activation and interfering with coagulation. Antiplatelet drugs, such as aspirin and clopidogrel or other ADP receptor inhibitors, which are particularly efficacious in combination, prevent stent thrombosis and recurrent ischemia in patients.77,78 Notwithstanding the bleeding, the success of these agents highlights the critical role of platelets.
Recent successful introduction of caplacizumab, an anti-VWF camelid antibody for treating thrombotic thrombocytopenic purpura,79 raises the possibility of targeting the VWF-GPIb axis to dampen biomaterial-platelet interactions. Other agents may include, for example, anti-GPIb inhibitors79 or N-acetylcysteine, which reduces the size of ULVWF multimers.80
Despite the introduction of low-molecular-weight heparins and direct oral anticoagulants that inhibit thrombin or FXa, heparin and warfarin have not been supplanted in preventing biomaterial-associated thrombosis. The situation may change, as new agents emerge and personalized dosage is optimized to achieve protection without bleeding.81 Currently, systemic heparin remains the first choice for preventing thrombosis on extracorporeal devices. Patients with ventricular assist devices, or mechanical or even bioprosthetic heart valves are still best managed with vitamin K antagonists to offset the risk of emboli formation and stroke.82
With the realization that the contact pathway participates in surface-induced thromboinflammation,83 there has been a surge in preclinical trials with inhibitors of FXIIa and/or FXIa, using peptides, proteins, aptamers, siRNAs, ASOs, synthetic compounds, and antibodies. Numerous preclinical and early-phase human trials have shown promise in preventing thromboinflammation with diverse biomaterials.84–86
Targeting complement to interfere with biomaterial-induced thromboinflammation is under consideration by several groups. In extracorporeal circulation models, blockades of C5a, C5b-9, or FD have dampened complement activation and reduced leukocyte and platelet activation87 and IL8 release.88 Compstatin analogues that target C3, were efficacious in an ex vivo model of hemodialysis, reducing TF expression and PMN activation, and in vivo in nonhuman primates.89 Anticomplement agents may prevent graft injury after organ transplantion.90 With more anticomplement agents, this strategy is likely to be more widely tested.20,91
As determinants of PMN recruitment, adhesion, and activation,55 β2-integrins are reasonable targets for diminishing their adverse effects. Heparin reduces leukocyte adhesion and activation, in part by binding to β2-integrin and blocking cell surface–expressed Mac-1 interactions with its ligands.92 Several antibodies and small molecules that target β2-integrins (eg, efalizumab and anti-M7) have been developed93–95; it is not known whether these agents prevent biomaterial-induced thromboinflammation.
Strategies to speed up PMN apoptosis may reduce biomaterial-induced thromboinflammation by modulating expression of pro- or antiapoptotic molecules96,97 or the stability of cyclin-dependent kinases. Several drugs and molecules, including aspirin, gasdermin D, annexin A1, and nicotinamide promote PMN apoptosis.98 Alternatively, PMNs may, in response to yet undefined cues, be induced to reverse direction and return to the circulation from inflammatory sites. Triggering “reverse migration” may be used to diminish biomaterial surface thromboinflammation. Finally, recent data indicate that, similar to macrophages, PMNs may be polarized to be pro- or anti-inflammatory.99 Delineating mechanisms to modulate PMN polarization may yield strategies to diminish the harmful consequences of biomaterial exposure to blood and promote regeneration.99
Local approaches
Many strategies have been used to enable biomaterials to resist protein adsorption.100 These include incorporating PEG, phosphatidycholine polymers, hyaluronic acid, zwitterionic molecules, hydrophilic polymers, or superhydrophobic modifications.11,98,101,102 Implants that are smooth and wettable, tend to induce lower foreign body reactions, protein adsorption, and cellular recruitment.61,103 Proteins such as albumin, layered with antibodies or ligand,104 may competitively reduce protein adsorption and adhesion or activation of platelets and leukocytes. Despite the limitations, such as susceptibility to destructive tissue remodelling, undesired breakdown products, and adverse adhesive properties, many such strategies are yielding promising results in preclinical models.101
Heparin has been used in vascular grafts and stents for decades105 and remains the mainstay. Heparin coatings reduce thromboinflammation.106 Unfractionated heparin, however, possesses inherent variable anticoagulant activity and a propensity to bind many proteins that interfere with that activity.107 Heparin-mimicking polyanions and thrombin inhibitors have thus been developed to reduce clotting and complement activation.108,109 Direct inhibition of coagulation has been explored by immobilizing thrombin inhibitors, such as hirudin, bivalirudin, and argatroban, or FXa inhibitors, such as fondaparinux. These protect against nonspecific protein adsorption and platelet activation in vitro in grafts and stents.110 The irreversible hirudin-thrombin interaction limits utility, but an analogue, bivalirudin, binds reversibly and is being explored for vascular grafts.111
NO reduces platelet adhesion, activation, and inflammation and promotes M2 polarization of macrophages. Ex vivo, heparin coimmobilized with an NO-generating catalyst, allows for stable, sustained and adjustable release and reduces platelet adhesion, coagulation, and inflammation.112 Other bioresponsive systems are being developed. For example, tPA loaded in a hydrogel was cross-linked with a thrombin-sensitive peptide, such that the tPA was released only when thrombin exceeded a threshold. Similarly, a self-regulated, thrombin-induced heparin release system has been devised (reviewed by Ippel and Dankers100). Such approaches may be extended to the complement system, to limit assembly and/or promote decay of C3 and C5 convertases via release of FH or soluble CD59.
Statins also exhibit antiplatelet and anti-inflammatory properties, blocking TXA2, inducing NO, reducing CD40L, and reducing MP release.113 In animal models, biodegradable vascular stents with rosuvastatin-loaded nanofibers, have exhibited reduced thromboinflammation with improved reendothelization.114
Other coatings include thrombomodulin,115 tPA, plasminogen,116,117 and corn trypsin inhibitor. FH or peptides with high affinity for FH, have been tested to restrict complement activation,118 as have modified polymers. Biomaterial additives to suppress NETosis, such as chloroquine diphosphate, are showing promise.59,119 Biodegradable scaffolds can also be used for sustained release of, for example, heparin, or dipyridamole.120,121
Biofunctional biomaterial surfaces that promote endothelialization are increasingly being developed122–124 and include, for example, surfaces that incorporate gene complexes that trigger endothelial proliferation and promote endothelial cell adhesion and proliferation over smooth muscle cells125 or peptides that stimulate endothelialization.100 Apyrase, an ADP-degrading enzyme, has been immobilized on materials coated with endothelial cells, as has heparin, the latter to recruit circulating AT.126
Advances in 3D printing and synthetic polymers127 are in parallel with the introduction of biodegradable scaffolds for vascular networks lined by endothelial cells that can accommodate dynamic blood flow. Endothelialized lumens can be supported by the desired parenchymal tissues seeded onto them. Such tissue engineering approaches, being tested in animal models, hold promise of a new generation of functional biomedical devices with biocompatible surfaces that better mimic the host.
Therapeutic cells
To overcome innate thromboinflammatory responses to transplanted or infused cells, systemic anticoagulation, anti-TF antibodies, activated protein C, and anti-inflammatory and antioxidant agents (eg, N-acetylcysteine, α-1 antitrypsin) have been tested.4,128 Heparin is routinely used for pancreatic islet cell transplantation,129 whereas low-molecular-weight dextran sulfate, which dampens complement, FXI, and FXII activation, was similarly effective in a phase 2 study.129 Modifying the surface of transplanted cells, with or without microencapsulating them with surface-modified biomaterials, are strategies that are also being pursued.130 For example, hepatocytes with peptide-conjugated PEG lipid, in conjunction with immobilized heparin conjugates,126 were exposed to blood in vitro. These surface-modified cells elicited less activation of platelets, coagulation, or complement. Cells have also been encapsulated in alginate microbeads or polymer coatings to limit immediate innate responses.131 Most promising are emerging ex vivo methodologies for preparing tissue-derived stem cells for transplantation that retain their properties while minimally eliciting an innate thromboinflammatory response.3
Taking advantage of thromboinflammation
We have limited this discussion to methods that prevent thromboinflammation. However, the host response to biomaterials may also be augmented to benefit. For example, to enhance antigen-specific immune responses for vaccination purposes, lymph node–targeting, complement-activating particles are being evaluated.132,133 Biomaterials designed to enhance clot formation are at various stages of use and development. These include, for example, hemostatic and wound-healing materials, self-propelling prothrombotic particles, and chitosan-based materials.134,135
Advancing the field
Host thromboinflammatory responses at the blood-biomaterial interface are complex, involving multiple dynamic and interactive cellular and molecular events (Figure 1). Although these pathways are well understood in health and disease, the translation of such information to the biomaterial interface design is in its infancy. There is a real gap between in vitro measurements and in vivo studies, especially at biomaterial surfaces136 where such interactions occur. Continuous in vivo measurements at the blood-biomaterial interface, of molecular and cellular events under appropriate biorheological conditions are lacking but critical for delineating relevant thromboinflammatory events. These measurements will inform which molecules or cells are being adsorbed or bound to the biomaterial and how they are interacting over time. They may be revealed partly with advanced microscopy technologies137 and are crucial for rational design of better biomaterials and their rapid personalized adaptation for different clinical purposes.
Currently used quantitative evaluations of responses to biomaterials that involve measuring activation products of different pathways (eg, coagulation, inflammation, and complement) yield valuable information, but they do not account for the complexity at the blood-biomaterial interface, nor are they likely to be sensitive enough to detect important low-level changes at the interface. Consideration should be given to incorporating surface-to-volume ratios when measuring soluble activation markers. Increasingly robust proteomics and genomics approaches are recommended to provide more sensitive localized measures of changes at the blood-biomaterial interface, as well as globally upon insertion of biomaterials. Such advanced technologies may provide a deeper understanding of the dynamic biomaterial-induced perturbation of homeostasis and the connectivity between the different players. In addition to identifying protein signatures on biomaterials in vivo,138 the information on the varying composition of the corona at the interface may provide insights that promote the design of innovative interventions to minimize or take advantage of thromboinflammatory responses to specific biomaterials.
Conclusion
The challenges of overcoming the innate thromboinflammatory response when blood meets a biomaterial, medical device, or cellular implant underscores the inherent power of the evolutionary forces that protect the host from invading organisms and wounds. Even with extraordinary advances in tissue engineering and biomaterial design, it is likely that we will continue to face these forces with new and creative strategies that will be built on a strong understanding of the underlying molecular and biochemical pathways that trigger and regulate the innate thromboinflammatory response at the blood-biomaterial interface.
Acknowledgments
Because of limited publication space, many works that were invaluable in the preparation of this review have not been cited as references. The authors apologize to all those who have contributed to this important area of research, yet could not be recognized.
E.M.C. was supported by operating grants from the Canadian Institutes of Health Research (CIHR) and the Natural Sciences and Engineering Research Council of Canada (NSERC). He holds a Tier 1 Canada Research Chair in Endothelial Cell Biology and is an adjunct Scientist with the Canadian Blood Services (CBS). J.N.K. acknowledges funding from CIHR and NSERC and received a Career Investigator Scholar award from the Michael Smith Foundation for Health Research (MSFHR).
Authorship
Contribution: E.M.C. and J.N.K. researched and wrote the manuscript and bear equal responsibility for its content.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Edward M. Conway, UBC Centre for Blood Research, 2350 Health Sciences Mall, LSC, 4th Floor, Vancouver, BC V6T 1Z3, Canada; e-mail: ed.conway@ubc.ca.


![The thromboinflammatory response of blood after exposure to a biomaterial surface. Although seemingly complex, this is a simplified representation of the multiple cellular and molecular interactions that occur when blood meets a biomaterial surface. Immediately upon contacting a biomaterial surface, plasma proteins (eg, fibrinogen, VWF, immunoglobulin [Ig], and complement proteins [C3, C3(H2O), C3b], FXII, and HK) are adsorbed. Complement is activated via all 3 pathways (AP, CP, and LP) as a result of deposition of C3 (C3, C3(H2O), C3b), complement-fixing Ig (recognized by C1q), and binding of MASPs. The activation of the plasma contact system, its key components being HK, PK, and FXII/FXIIa, results in generation of thrombin (IIa) via the intrinsic pathway, as well as PKa, both of which amplify the thromboinflammatory response via several pathways. Platelets rapidly adhere to adsorbed fibrinogen, augmented under high shear rate, by VWF. These are activated by the intrinsic pathway-generated thrombin and C3a and C5a, that also exert proinflammatory and prothrombotic effects on all immune cells via their respective cell-surface receptors (Figure 3). Cytokines and chemokines are released from the platelets and recruit inflammatory leukocytes, which themselves then release more cytokines and chemokines. C5a and C5b-9 (MAC) activate TF on the monocytes/macrophages which were recruited by chemokines that are released by activated platelets and neutrophils, the latter of which may undergo NETosis, thereby further promoting local and systemic inflammation, thrombin generation, and clot formation. Thrombin, generated via the intrinsic and extrinsic (TF-dependent) pathways, also triggers inflammatory responses via PAR signaling on all immune cells. On the right side, adjacent host vascular endothelium with underlying smooth muscle cells, is shown to highlight how its integrity may be jeopardized during the innate thromboinflammatory response.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/13/10.1182_blood.2020007209/5/m_bloodbld2020007209cf1.png?Expires=1767773505&Signature=d3-yjX8yIYU2xRaunC4CfUAEwHmfhxkinv0eLcN2lU4WvasuS95oPrxVxj1kNQf7WaDH-JNjd3Aqo8j4nytEgEIsh33r1gOSedMzj-Thc3eqIrlGenTSzDlkVxi7MbDBwRqlcQUiQBODeKzaSGQHBBxwPXfz8GheZGtrj7weLy8q0pNECBXavRahale2SsuIMcbRYomAJdn2-abXvzN~xqKgcmH7njlWee9cTiEhYBffQAzvfKUZ0M~ThKlMdGExb0PoHkWOI9SO7tyBDTY~s6~-Hn0jRjXAjtAM-e7IKLY6y0iYU5Ybo40NZFmSxXLLsKPG8b4m0RWNTImHGddBvA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)