

TO THE EDITOR:
Diffuse large B-cell lymphoma (DLBCL) is the most frequent lymphoid malignancy in adults. Recently, landmark multi-omic studies have provided comprehensive collections of molecular alterations in more than 1800 DLBCL tumors (Reddy et al,1 N = 1001; Schmitz et al,2 N = 574; Chapuy et al,3 N = 304). We recently reported that BCL7A, a member of the SWItch/Sucrose Non-Fermentable chromatin remodeling complex and a tumor suppressor gene in DLBCL, is recurrently mutated at its first splice donor site in DLBCL.4 Although these splice site mutations impaired the function of BCL7A, they had been overlooked by large-scale studies.4 Importantly, Reddy et al1 provided the largest whole-exome sequencing dataset to date in DLBCL (N = 1001), but they did not analyze splice sites. Based on our experience, we wondered if other genes undergo recurrent but overlooked splice site mutations in DLBCL.
To identify previously missed splice site mutations in the dataset of Reddy et al,1 we performed unpaired variant calling at splice sites followed by strict filtering using the somatic mutation data from Schmitz et al2 and Chapuy et al3 (supplemental Methods and supplemental File 1 available on the Blood Web site). We found 29 genes that had likely somatic splice site mutations in at least 5 patients from the cohort of Reddy et al1 (Figure 1A; supplemental File 2). Remarkably, the mutation frequency per nucleotide in these genes was a median of ∼8 times higher at splice sites than at coding sequences (Figure 1B; supplemental File 3). The accumulation of mutations at splice sites affected known DLBCL genes, such as BCL7A (19 times), SGK1 (12 times), CD79B (9 times), and BCL6 (9 times). The inclusion of splice site mutations increased the mutation frequency of BCL7A by ∼44%, of SGK1 by ∼22%, of CD79B by ∼32%, and of BCL6 by ∼18%. The mutation frequency of BCL7A was comparable to that from our previous report.4 Our analysis also revealed novel genes that were not reported as mutated in the coding sequence by Reddy et al,1 including ZFP36L1, POU2AF1, GRHPR, PABPC1, CD74, LAPTM5, and MYO1E. Interestingly, ZFP36L1 had a mutation frequency of ∼10% in the other 2 analyzed datasets (∼3%-4% if only considering splice site mutations), and it has been proposed as a tumor suppressor gene in germinal center–derived B-cell lymphomas.5 The splice site mutation frequencies of our 29 selected genes in the dataset of Reddy et al1 correlated moderately with those from Schmitz et al2 (Kendall τ = 0.28, P = .04) and Chapuy et al3 (Kendall τ = 0.44, P = .002). Because of the lack of public sequencing data from normal samples, when we reanalyzed the cohort of Reddy et al,1 we applied conservative filters that probably excluded genuine somatic mutations (supplemental Methods). We expect that an analysis that incorporates information from matched normal samples may reveal even more splice site mutant genes.
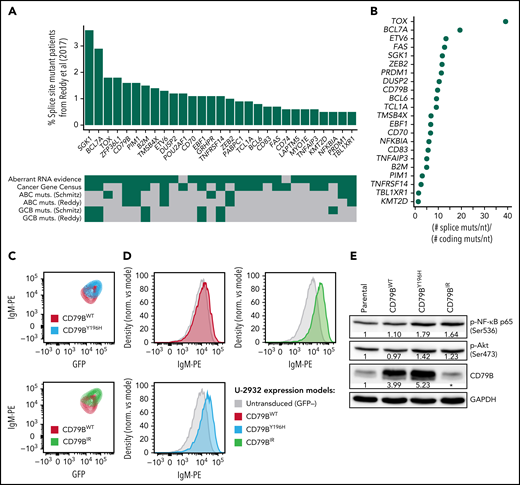
Recurrent splice site mutations in DLBCL. (A) Genes recurrently mutated at splice sites according to our reanalysis of the dataset of Reddy et al1 (N = 1001). We only considered mutations that were confirmed as somatic by Schmitz et al2 or Chapuy et al.3 The genes shown in the figure were mutated in at least 5 patients. For each gene, the frequency of splice site mutant patients is shown in the vertical axis. The heatmap below shows the presence (green) or absence (gray) of relevant features for each gene. Aberrant RNA evidence, at least 1 splice site mutant patient had an RNA aberration that could be attributed to the mutation according to our analyses; Cancer Gene Census, the gene is part of the Cancer Gene Census; GCB-/ABC muts., the gene is preferentially mutated in the GCB or ABC subtype according to Reddy et al1 or Schmitz et al.2 (B) Ratio between the frequency of splice site mutations per nucleotide and the frequency of coding mutations per nucleotide for the 22 recurrent splice site mutant genes that had nonzero coding mutation frequency in the dataset of Reddy et al.1 (C) Mutant CD79BIR increases surface immunoglobulin M (IgM) at a higher extent than CD79BY196H. The ABC DLBCL line U-2932 was transduced with CD79BWT, mutant CD79BY196H, or mutant CD79BIR. Surface IgM is depicted gating on a cotransduced green fluorescent protein (GFP) marker to identify the subset of transduced cells with equivalent ectopic CD79B RNA expression. (D) Surface IgM expression in U-2932 cells transduced with the indicated CD79B isoforms. U-2932 GFP-positive transduced cells are compared with untransduced cells. (E) Western blot of CD79B, phosphorylated RELA/p65 (Ser536), and phosphorylated Akt (Ser473) in U-2932 cells after overexpression of mutant or wild-type CD79B. The numbers indicate the fold change between each CD79B overexpression model and the parental cell line, previously normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH), according to a densitometry analysis using ImageJ. *The epitope recognized by the anti-CD79B antibody is within the region that is lost by CD79BIR, which is why overexpression cannot be detected by this method. However, we confirmed CD79BIR plasmid overexpression by flow cytometry (GFP+ signal).
Recurrent splice site mutations in DLBCL. (A) Genes recurrently mutated at splice sites according to our reanalysis of the dataset of Reddy et al1 (N = 1001). We only considered mutations that were confirmed as somatic by Schmitz et al2 or Chapuy et al.3 The genes shown in the figure were mutated in at least 5 patients. For each gene, the frequency of splice site mutant patients is shown in the vertical axis. The heatmap below shows the presence (green) or absence (gray) of relevant features for each gene. Aberrant RNA evidence, at least 1 splice site mutant patient had an RNA aberration that could be attributed to the mutation according to our analyses; Cancer Gene Census, the gene is part of the Cancer Gene Census; GCB-/ABC muts., the gene is preferentially mutated in the GCB or ABC subtype according to Reddy et al1 or Schmitz et al.2 (B) Ratio between the frequency of splice site mutations per nucleotide and the frequency of coding mutations per nucleotide for the 22 recurrent splice site mutant genes that had nonzero coding mutation frequency in the dataset of Reddy et al.1 (C) Mutant CD79BIR increases surface immunoglobulin M (IgM) at a higher extent than CD79BY196H. The ABC DLBCL line U-2932 was transduced with CD79BWT, mutant CD79BY196H, or mutant CD79BIR. Surface IgM is depicted gating on a cotransduced green fluorescent protein (GFP) marker to identify the subset of transduced cells with equivalent ectopic CD79B RNA expression. (D) Surface IgM expression in U-2932 cells transduced with the indicated CD79B isoforms. U-2932 GFP-positive transduced cells are compared with untransduced cells. (E) Western blot of CD79B, phosphorylated RELA/p65 (Ser536), and phosphorylated Akt (Ser473) in U-2932 cells after overexpression of mutant or wild-type CD79B. The numbers indicate the fold change between each CD79B overexpression model and the parental cell line, previously normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH), according to a densitometry analysis using ImageJ. *The epitope recognized by the anti-CD79B antibody is within the region that is lost by CD79BIR, which is why overexpression cannot be detected by this method. However, we confirmed CD79BIR plasmid overexpression by flow cytometry (GFP+ signal).
To evaluate the significance of our findings, we analyzed clinically relevant features in the 29 recurrent splice site mutant genes (Figure 1A). Of the 29 genes, 18 (62.1%) were known cancer genes according to the Cancer Gene Census (CGC).6 They included SGK1, BCL7A, CD79B, and PIM1, among others. Three of the CGC genes had not been reported by Reddy et al1: POU2AF1, PABPC1, and CD74. POU2AF1 is involved in the formation of germinal centers in mice, and splice site mutations in this gene may be related to the transformation of follicular lymphoma to DLBCL.7,8 In addition, the 29 selected genes included 5 of the 12 (42%) genes whose mutations are specific to the germinal center B-cell-like (GCB) subtype and 5 of the 8 (63%) genes whose mutations are specific to the activated B-cell-like (ABC) subtype according to Reddy et al.1 Furthermore, exonic mutations and RNA-altering splice site mutations (see below) in CD79B and PIM1 were associated with survival in both cohorts of Reddy et al1 and Schmitz et al2 (supplemental File 4). Taken together, these results highlight the clinical relevance of splice site mutant genes.
Next, we evaluated whether splice site mutations altered the splicing of the affected RNAs using the RNA sequencing (RNA-Seq) data from Reddy et al1 and Schmitz et al.2 We used the MAJIQ tool followed by manual curation (supplemental Methods).9 In 27 of 29 (93%) genes, at least 1 splice site mutant patient had an RNA aberration (supplemental Files 5 and 6). Furthermore, in 14 of 29 genes (48%), at least half of the splice site mutant samples in both datasets had RNA aberrations. The most frequent RNA aberration according to a sample-by-sample analysis was intron retention (110 cases), followed by expression of the mutant splice site along with a few intronic nucleotides (94 cases), cryptic splice sites (92 cases), exon skipping (54 cases), and increased use of alternative canonical isoforms (49 cases; Figure 2; supplemental Figures 1-5). In BCL7A, mutations in the first splice donor site often led to the use of a cryptic splice donor site in exon 1 that resulted in a loss of 27 amino acids, as we previously reported.4 Overall, RNA sequencing data have allowed us to confirm the impact of splice site mutations in most affected genes.
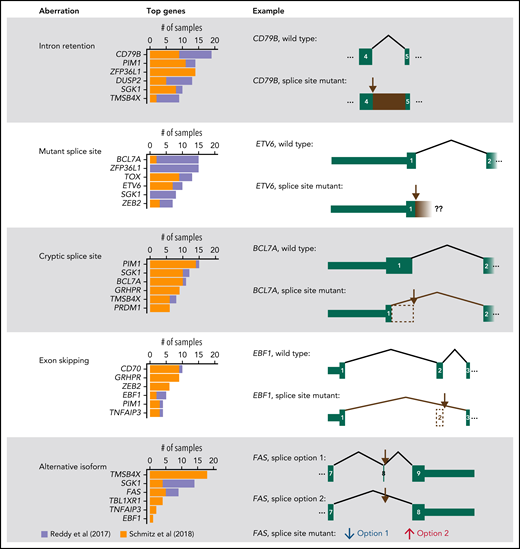
Summary of RNA aberrations identified in splice site mutant genes. For each type of aberration, the top 6 most affected genes are shown. In addition, 1 illustrative example is represented schematically. Brown arrows mark the most recurrently mutated splice site in the gene. Exon numbering was based on the following transcripts from Ensembl v28: ENST00000006750.7 (CD79B), ENST00000396373.8 (ETV6), ENST00000261822.4 (BCL7A), ENST00000313708.10 (EBF1), ENST00000355740.6 (FAS, splice option 1), and ENST00000355279.2 (FAS, splice option 2). In FAS, splice site mutant samples have decreased use of “splice option 1” and increased use of “splice option 2.” Our analysis estimates the differential use of each splice junction and not the expression of whole transcripts.
Summary of RNA aberrations identified in splice site mutant genes. For each type of aberration, the top 6 most affected genes are shown. In addition, 1 illustrative example is represented schematically. Brown arrows mark the most recurrently mutated splice site in the gene. Exon numbering was based on the following transcripts from Ensembl v28: ENST00000006750.7 (CD79B), ENST00000396373.8 (ETV6), ENST00000261822.4 (BCL7A), ENST00000313708.10 (EBF1), ENST00000355740.6 (FAS, splice option 1), and ENST00000355279.2 (FAS, splice option 2). In FAS, splice site mutant samples have decreased use of “splice option 1” and increased use of “splice option 2.” Our analysis estimates the differential use of each splice junction and not the expression of whole transcripts.
The most frequent RNA aberration affected CD79B (Figure 2; supplemental Figure 6), which accumulated 23 mutations in its fourth splice donor site, out of which at least 18 caused retention of intron 4 (CD79BIR). The retained intron introduced a premature stop codon just before the immunoreceptor tyrosine-based activation motif (ITAM)-containing domain. CD79B and CD79A form dimers that, together with immunoglobulins at the B-cell membrane, constitute B-cell receptors (BCRs).10 The ITAMs of CD79A and CD79B are involved in BCR signaling and internalization. In DLBCL, the ITAMs of CD79A and, most frequently, CD79B recurrently undergo point mutations and deletions that prevent BCR internalization, increasing surface BCR levels and causing overactive oncogenic BCR signaling.10,11 Indeed, when we overexpressed the most frequent CD79BIR variant in U-2932 and Ri-1 (Riva) cells, surface BCR levels increased compared with overexpression of wild-type CD79B or of the most frequent exonic mutation, CD79BY196H (Figure 1C-D; supplemental Figures 7 and 8). Furthermore, CD79BIR and CD79BY196H, but not CD79BWT, increased phosphorylation of AKT and RELA/p65 (Figure 1E displays the Western blot for U-2932 cells), suggesting an increase in oncogenic signaling via AKT and nuclear factor-κB (Figure 1E). Importantly, mutations in CD79B, together with MYD88L265P, define the “MCD” clinical subgroup, which is characterized by a poor prognosis but good response to the Bruton's tyrosine kinase inhibitor ibrutinib.2,11 Primary central nervous system lymphomas harboring CD79BY196 and CD79B splice site mutations show similar responses to ibrutinib, which agrees with them having similar functional consequences.12 In addition, mutations at the fourth splice acceptor site of CD79B may also cause RNA aberrations,13 but these mutations were rare in our analyzed datasets. Taken together, our results highlight the functional relevance of splice site mutations in CD79B in DLBCL.
In conclusion, splice site mutations recurrently affect key DLBCL genes, such as those related with disease subtype or with patient outcome. In particular, mutations in the fourth splice donor site of CD79B increase surface BCR similar to the well-known oncogenic CD79BY196 mutations. Splice site mutations can have important clinical applications. Splice site mutant genes may be targeted by anticancer drugs, such as the recently US Food and Drug Administration–approved capmatinib, which targets MET exon 14 skipping in metastatic non–small-cell lung cancer.14,15 Furthermore, RNA aberrations caused by splice site mutations may generate neoepitopes for immunotherapy.16 Therefore, splice site mutations may be a major source of clinically relevant alterations in cancer.
Acknowledgments
P.P.M.’s laboratory is funded by Aula de Investigación sobre la Leucemia infantil: Héroes contra la Leucemia, the Ministry of Economy of Spain (grant SAF2015-67919-R), Junta de Andalucía (grants PIGE-0440-2019, PI-0135-2020, and P20_00688), University of Granada (grants B-CTS-126-UGR18, B-CTS-480-UGR20, and E-CTS-304-UGR20), and the Spanish Association for Cancer Research (LABORATORY-AECC-2018). A.A. was supported by a FPU17/00067 fellowship (Spanish Ministry of Science, Innovation and Universities). J.R.P.-M. was supported by a FPU18/03709 fellowship (Spanish Ministry of Science, Innovation and Universities). J.C.Á.-P. was supported by a Marie Sklodowska Curie Actions postdoctoral fellowship (H2020-MSCA-IF-2018).
Data from Reddy et al1 can be accessed at the European Genome Phenome Archive (EGA dataset EGAD00001003600). The results shown here are in part based on data generated by the National Cancer Institute’s Clinical Trials Sequence Program (dbGaP ID phs001175.v1.p1), the “Genomic Variation in Diffuse Large B Cell Lymphomas” project (dbGaP ID phs001444.v1.p1), and The Cancer Genome Atlas (dbGaP ID: phs000178.v11.p8).
Authorship
Contribution: P.P.M. conceived the study, coordinated the scientific team, and allocated the funding for the project; A.A. performed the computational analyses and wrote the first draft of the manuscript; J.C.Á.-P. performed the laboratory experiments related to CD79B; and all authors discussed, reviewed, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for C.B.-G. is Institut Curie, Paris Sciences et Lettres Research University, Sorbonne University, INSERM U934/CNRS UMR3215, Paris, France.
Correspondence: Pedro P. Medina, Department of Biochemistry and Molecular Biology I, Faculty of Sciences, University of Granada, Avenida de Fuente Nueva, s/n, 18071 Granada, Spain; e-mail: pedromedina@ugr.es.
Requests for data sharing may be submitted to Pedro P. Medina (pedromedina@ugr.es).
The online version of this article contains a data supplement.

