

Key Point
T-cell expression of GPR109A is necessary for metabolic function and alloreactivity.

Abstract
The intestinal microbiota is essential for the fermentation of dietary fiber into short-chain fatty acids (SCFA) such as butyrate, acetate, and propionate. SCFAs can bind to the G-protein-coupled receptors GPR43 and GPR109A (HCAR2), with varying affinities to promote cellular effects in metabolism or changes in immune function. We explored the role of GPR109A as the main receptor for butyrate in mouse models of allogeneic hematopoietic cell transplantation (allo-HCT) and graft-versus-host disease (GVHD). Deletion of GPR109A in allo-HCT recipients did not affect GVHD, but transplantation of T cells from GPR109A knockout (KO) (Gpr109a−/−) mice into allo-HCT recipient mice significantly reduced GVHD morbidity and mortality compared with recipients of wild-type (WT) T cells. Recipients of Gpr109a−/− T cells exhibited less GVHD-associated target organ pathology and decreased proliferation and homing of alloreactive T cells to target tissues. Although Gpr109a−/− T cells did not exhibit immune deficits at a steady state, following allo-activation, Gpr109a−/− T cells underwent increased apoptosis and were impaired mitochondrial oxidative phosphorylation, which was reversible through antioxidant treatment with N-acetylcysteine (NAC). In conclusion, we found that GPR109A expression by allo-activated T cells is essential for metabolic homeostasis and expansion, which are necessary features to induce GVHD after allo-HCT.
Introduction
The intestinal microbiota consists of trillions of bacteria, most of which are nonpathogenic commensals important for maintaining intestinal homeostasis. One microbial function that is beneficial to the host is the fermentation of nondigestible carbohydrates into metabolites, notably the short-chain fatty acids (SCFA) acetate, propionate, and butyrate. SCFAs bind to specific G-protein-coupled receptors GPR41, GPR43, and GPR109A (encoded by Ffar2, Ffar3, and Hcar2, respectively) on various cell types to regulate intestinal homeostasis.1,2 SCFA uptake can also occur through passive diffusion across the membrane or active transport via SLC5a8.3 SCFAs play a significant role in promoting and maintaining mucosal homeostasis.4-7 For example, butyrate can enhance the protective intestinal mucus layer by inducing MUC-2 expression in epithelial cells,8 and mice lacking either GPR43 or GPR109A are more susceptible to chemically induced colitis.9,10 Butyrate also inhibits histone deacetylase (HDAC) activity in dendritic cells to induce differentiation of regulatory T cells, or it can act directly on CD4+ T cells in a GPR43-dependent manner to enhance regulatory T-cell differentiation.11,12
Recent studies by our group and others have demonstrated the importance of the intestinal microbiota and its metabolites in the pathophysiology of graft-versus-host disease (GVHD) following allogeneic hematopoietic cell transplantation (allo-HCT).13-17 Allo-HCT is a therapy for hematologic malignancies administered with the intent to cure. Still, nearly 50% of patients develop GVHD, an inflammatory and often lethal syndrome primarily mediated by alloreactive donor T cells. Acute GVHD predominantly affects the intestines, skin, and liver, and patients with lower gastrointestinal tract involvement are at the highest risk of mortality.
GVHD is accompanied by marked changes in the composition of intestinal microbes and their associated metabolites, such as SCFAs.18-21 In the context of GVHD, we recently reported that propionate and, to a lesser extent, butyrate, activates GPR43 expression on IECs, leading to activation of the NLRP3 inflammasome and downstream IL-18 expression, which promotes maintenance of intestinal epithelial barrier function resulting in a reduction of GVHD in mice.22,23 In another study, we demonstrated that butyrate could mitigate GVHD in a GPR43-independent manner by maintaining enterocyte homeostasis and restoring IEC junction integrity after allo-HCT-induced mucosal injury.16
The role of GPR109A, which is the primary butyrate receptor and also binds the B vitamin niacin, has not yet been explored in GVHD. Therefore, we hypothesized that GPR109A may play a role in the pathophysiology of acute GVHD.
Materials and methods
Mice and bone marrow transplantation and assessment of GVHD
Gpr109a−/− mice, a gift from Klaus Pfeffer (University of Dusseldorf), were backcrossed onto the C57BL/6 (B6) (CD45.2 B6, H-2b) background for ≥10 generations. Wild type (WT) mice (GPR109a+/+) were generated by crossing Gpr109a+/− to generate littermates. WT C57BL/6 (where indicated), BALB/c, and 129S1/SvImJ mice were obtained from Jackson Laboratory. All mice used for experiments were 6 to 9 weeks old. Mouse allo-HCT experiments were performed as previously described, and in all transplants bone marrow (BM) was T-cell-depleted prior to transplant.24,25 For the A20 tumor experiment, when mice died, we performed an autopsy and analyzed the liver for the presence of tumors, weighed the spleen, and submitted for pathology to confirm a tumor death. If no tumor was detected, the cause of death was counted as due to GVHD. Mice were monitored for survival and clinical GVHD symptoms26 or were killed for blinded histopathologic and flow cytometric analysis, as described.27 Experiments were conducted in compliance with the institutional guidelines at MSKCC.
Flow cytometry and cell sorting
Cells were stained for surface markers with antibody mixtures for 20 minutes at 4°C, followed by intracellular staining with the fixation/permeabilization kit (eBioscience) per the manufacturer’s instructions. All flow cytometry was performed on a FACS Symphony A5 (BD Biosciences) and analyzed with FlowJo (Tree Star Software). Cell sorting was performed on an Aria II cytometer (BD Biosciences), and cell populations were sorted to >95% purity.
Statistical analyses
Data were processed in GraphPad Prism 5.0 software. Statistical comparisons between 2 groups were performed with the nonparametric unpaired Mann-Whitney U test. Survival data were analyzed with the Mantel-Cox log-rank test. P values <.05 were considered statistically significant. All data shown in graphs represent the mean ± standard deviation (SD) of each group.
More details on the methods used are presented in the supplemental Material.
Data sharing
16S rRNA sequencing data can be found at accession number: PRJNA765037.
RNAseq data can be found at accession number GSE185903.
Results
GPR109A-deficient donor T cells induce less GVHD morbidity and mortality
We have previously demonstrated that GPR43 signaling in nonhematopoietic tissues of the host protects against intestinal GVHD.23 In contrast to GPR43, which preferentially binds to propionate and acetate, GPR109A has specificity for binding to butyrate. Luminal butyrate supports mucosal integrity after allo-HCT16; therefore, we hypothesized that GRP109a signaling would be involved in the pathogenesis of GVHD.
We first examined the role of GPR109A in allo-HCT recipient tissues by transplanting WT grafts into Gpr109a−/− knockout (KO) or WT recipient mice following a period of cohousing to equalize the composition of the intestinal microbiota (supplemental Figure S1A). In a major histocompatibility complex (MHC)-matched, minor histocompatibility antigen mismatched model of GVHD, lethally irradiated WT or KO recipients, both bred on a C57BL/6 [B6] inbred mouse strain background, were transplanted with either LP/J BM only (BM; for non-GVHD controls) or BM with purified splenic T cells (BM + T; for GVHD induction). No difference in survival or clinical GVHD scores was observed between WT and KO recipient mice (Figure 1A).
![GPR109A−/− T cells cause less GVHD than WT T cells. (A) Survival and clinical GVHD scores of lethally irradiated C57BL/6 (B6) or Gpr109a−/− recipients transplanted with 5 × 106 T cell-depleted bone marrow (BM) cells and 1 × 106 LP/J splenic T cells (LP BM + T). Controls were transplanted with T cell-depleted BM only (LP BM). (B) Survival and clinical GVHD scores of lethally irradiated BALB/c mice transplanted with 5 × 106 T cell-depleted BM cells and 1 × 106 WT T cells (B6 + B6 T cells), WT BM with Gpr109a−/− T cells (B6 BM + Gpr109a−/− T), or Gpr109a−/− BM with WT T cells (Gpr109a−/− BM + B6 T). Controls were transplanted with T cell-depleted BM only (B6 BM) or Gpr109a−/− BM only (Gpr109a−/− BM) in a major (B6 into BALB/c) MHC-mismatched model of GVHD. (C) Same as in B, in a minor antigen mismatched, MHC-matched model of GVHD (B6 into 129S1). (D) BM chimeras, WT B6 Ly5.2 into Ly5.1 or Gpr109a−/− Ly5.2 into Ly5.1 were used as donors. Survival and clinical GVHD scores of lethally irradiated BALB/c mice transplanted with 5 × 106 WT T cell-depleted BM cells and 1 × 106 WT T cells ([B6 → Ly5.1] BM + [B6 → Ly5.1] T) or WT T cell-depleted BM with Gpr109a−/− T cells ([B6 → Ly5.1] BM + [Gpr109a−/− → Ly5.1] T). Controls were transplanted with WT ([B6 → Ly5.1] BM) or Gpr109a−/− T cell-depleted BM ([Gpr109a−/− → Ly5.1]) only. (E) Lethally irradiated BALB/c mice transplanted with 5 × 106 WT T cell-depleted BM cells, 1 × 106 A20 tumor cells, and 0.5 × 106 WT (B6 BM + B6 T + A20) or Gpr109a−/− T cells (B6 BM + Gpr109a−/− T + A20). Controls were transplanted with WT T cell-depleted BM and A20 tumor cells (B6 BM + A20) only. Cause of death is plotted in the table to the right. Comparisons of groups for survival curves in (A) to (E) were performed by Mantel-Cox log rank test for survival. For GVHD scores, data represent the mean ± standard error, comparison of groups performed by two-way ANOVA. All results from two to three independent experiments.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/15/10.1182_blood.2021010719/4/m_bloodbld2021010719f1.png?Expires=1769295104&Signature=jK~pCTTHdYEnbd2AejHS4E9OqIy0D6TxKcLYXB4DNYVSemM9podZNwnFNf~cbPb3MV9i9d3MF84HVTCnR5s20N5IawOozNkulciryjHvpFEC7okTMy-duo5gH2i7w~b02noVOFSUPHb3wNzi1BXsRdhdhJq3YvFRRfxA8M5ePeIkIeTBzSTm~Mhp04HC3V61uNomrkU2TkvIyum1BMpHbL-XjSNjJJonCskUGaNfHZkbKln2UmjL1HqqTKrpZrFW9GRBSm8~S16TijVbDn8KUzMmBHKwwl8V-y-4JqxBM0TQmOIctY4MhlWPew5UPDm~6XY2BpwiwTjXK67qEtswQA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
GPR109A−/− T cells cause less GVHD than WT T cells. (A) Survival and clinical GVHD scores of lethally irradiated C57BL/6 (B6) or Gpr109a−/− recipients transplanted with 5 × 106 T cell-depleted bone marrow (BM) cells and 1 × 106 LP/J splenic T cells (LP BM + T). Controls were transplanted with T cell-depleted BM only (LP BM). (B) Survival and clinical GVHD scores of lethally irradiated BALB/c mice transplanted with 5 × 106 T cell-depleted BM cells and 1 × 106 WT T cells (B6 + B6 T cells), WT BM with Gpr109a−/− T cells (B6 BM + Gpr109a−/− T), or Gpr109a−/− BM with WT T cells (Gpr109a−/− BM + B6 T). Controls were transplanted with T cell-depleted BM only (B6 BM) or Gpr109a−/− BM only (Gpr109a−/− BM) in a major (B6 into BALB/c) MHC-mismatched model of GVHD. (C) Same as in B, in a minor antigen mismatched, MHC-matched model of GVHD (B6 into 129S1). (D) BM chimeras, WT B6 Ly5.2 into Ly5.1 or Gpr109a−/− Ly5.2 into Ly5.1 were used as donors. Survival and clinical GVHD scores of lethally irradiated BALB/c mice transplanted with 5 × 106 WT T cell-depleted BM cells and 1 × 106 WT T cells ([B6 → Ly5.1] BM + [B6 → Ly5.1] T) or WT T cell-depleted BM with Gpr109a−/− T cells ([B6 → Ly5.1] BM + [Gpr109a−/− → Ly5.1] T). Controls were transplanted with WT ([B6 → Ly5.1] BM) or Gpr109a−/− T cell-depleted BM ([Gpr109a−/− → Ly5.1]) only. (E) Lethally irradiated BALB/c mice transplanted with 5 × 106 WT T cell-depleted BM cells, 1 × 106 A20 tumor cells, and 0.5 × 106 WT (B6 BM + B6 T + A20) or Gpr109a−/− T cells (B6 BM + Gpr109a−/− T + A20). Controls were transplanted with WT T cell-depleted BM and A20 tumor cells (B6 BM + A20) only. Cause of death is plotted in the table to the right. Comparisons of groups for survival curves in (A) to (E) were performed by Mantel-Cox log rank test for survival. For GVHD scores, data represent the mean ± standard error, comparison of groups performed by two-way ANOVA. All results from two to three independent experiments.
GPR109A−/− T cells cause less GVHD than WT T cells. (A) Survival and clinical GVHD scores of lethally irradiated C57BL/6 (B6) or Gpr109a−/− recipients transplanted with 5 × 106 T cell-depleted bone marrow (BM) cells and 1 × 106 LP/J splenic T cells (LP BM + T). Controls were transplanted with T cell-depleted BM only (LP BM). (B) Survival and clinical GVHD scores of lethally irradiated BALB/c mice transplanted with 5 × 106 T cell-depleted BM cells and 1 × 106 WT T cells (B6 + B6 T cells), WT BM with Gpr109a−/− T cells (B6 BM + Gpr109a−/− T), or Gpr109a−/− BM with WT T cells (Gpr109a−/− BM + B6 T). Controls were transplanted with T cell-depleted BM only (B6 BM) or Gpr109a−/− BM only (Gpr109a−/− BM) in a major (B6 into BALB/c) MHC-mismatched model of GVHD. (C) Same as in B, in a minor antigen mismatched, MHC-matched model of GVHD (B6 into 129S1). (D) BM chimeras, WT B6 Ly5.2 into Ly5.1 or Gpr109a−/− Ly5.2 into Ly5.1 were used as donors. Survival and clinical GVHD scores of lethally irradiated BALB/c mice transplanted with 5 × 106 WT T cell-depleted BM cells and 1 × 106 WT T cells ([B6 → Ly5.1] BM + [B6 → Ly5.1] T) or WT T cell-depleted BM with Gpr109a−/− T cells ([B6 → Ly5.1] BM + [Gpr109a−/− → Ly5.1] T). Controls were transplanted with WT ([B6 → Ly5.1] BM) or Gpr109a−/− T cell-depleted BM ([Gpr109a−/− → Ly5.1]) only. (E) Lethally irradiated BALB/c mice transplanted with 5 × 106 WT T cell-depleted BM cells, 1 × 106 A20 tumor cells, and 0.5 × 106 WT (B6 BM + B6 T + A20) or Gpr109a−/− T cells (B6 BM + Gpr109a−/− T + A20). Controls were transplanted with WT T cell-depleted BM and A20 tumor cells (B6 BM + A20) only. Cause of death is plotted in the table to the right. Comparisons of groups for survival curves in (A) to (E) were performed by Mantel-Cox log rank test for survival. For GVHD scores, data represent the mean ± standard error, comparison of groups performed by two-way ANOVA. All results from two to three independent experiments.
We next explored the role of GPR109A in donor allo-HCT grafts by transplanting grafts from either WT B6 or KO donor mice into both MHC-disparate (B6/KO to BALB/c) or MHC-matched (B6/KO to 129S1) recipients to induce acute GVHD. Recipients of WT BM with KO T cells exhibited significantly reduced GVHD compared with recipients of WT BM with WT T cells or KO BM with WT T cells (Figure 1B-C). In these experiments, we used KO mice bred at our institution and WT mice purchased from Jackson Laboratories, which raises the possibility of a mouse batch effect due to genetic or animal housing differences. To exclude this confounder, we repeated these experiments with donor T cells from WT and KO littermates and observed again the significant difference in GVHD-related mortality in recipients receiving KO vs WT T cells (supplemental Figure 1B). When transplanting mice with both KO BM and KO T cells, we still observed significantly improved survival in KO cell recipients compared with mice transplanted with WT BM and T cells (supplemental Figure 1C). We conclude that donor T cells deficient for GPR109A exhibit less alloreactivity in GVHD models.
To exclude the possibility that the GPR109A-deficient environment during T cell development was inducing changes in the KO donor T cells, we generated BM chimera mice to be used as donors. Specifically, we transplanted BM cells from WT Ly5.2 or GPR109a−/− Ly5.2 mice into lethally irradiated WT Ly5.1 mice and allowed them to reconstitute to achieve 75% to 90% chimerism (supplemental Figure 1D). We used these chimeric mice as donors in the MHC-disparate GVHD model (B6 to BALB/c). We found that KO T cells that differentiated in a WT chimera before transplantation induced less GVHD compared with mice that received WT BM with WT T cells (Figure 1D). These results suggested that the ability of GPR109A to affect alloreactivity is intrinsic to the donor T cells. Of note, we did not observe a similar effect when transplanting WT BM with T cells from Gpr43−/− mice (supplemental Figure 1E) into BALB/c recipients.
To examine whether KO T cells are still capable of eliciting an antitumor response, we used a graft vs tumor (GVT) model in which B6 BM and T cells were transplanted into BALB/c recipients that also received A20 B cell lymphoma cells (Figure 1E). Recipients of KO T cells showed attenuated GVHD and no increase in deaths due to lymphoma (Figure 1E table inset). We conclude that the absence of GPR109A in donor T cells does not impair GVT activity against A20.
We further compared the ability of WT vs KO T cells to respond to a mouse cytomegalovirus (MCMV) infection by generating mixed BM chimeras with WT and KO BM, then subsequently infected these mice with MCMV (supplemental Figure 2A-B). We observed minimal differences in frequency of CD4+ and CD8+ T cells postinfection (supplemental Figure 2C-I). These results suggest that KO T cells retain the ability to mount antiviral responses despite a marked reduction in capacity to induce lethal acute GVHD.
Activated T cells express GPR109A, and the absence of GPR109A does not alter the T-cell phenotype
Having identified a T cell-intrinsic role for GPR109A in conferring GVHD activity, we sought to confirm that GPR109A expression could be detected in T cells. GPR109A is primarily expressed on the colonic epithelium, adipose tissue, and immune cells such as dendritic cells and macrophages.28-30 Splenic T cells, B cells, and NK cells have been reported to express low or undetectable levels of Gpr109a transcripts at steady state.31,32 We found that unstimulated, naïve CD4+ and CD8+ T cells express Gpr109a transcripts at low levels compared with positive control (F4-80+ macrophages), but the expression of Gpr109a transcripts, as well as GPR109A protein expression in T cells, was significantly increased upon 72 hours of in vitro T-cell receptor activation with anti-CD3/CD28 (Figure 2A-B). To determine at what point the expression of GPR109A is highest after in vivo allo-activation, we transplanted WT BM and WT T cells into BALB/c recipient mice and FACS-sorted CD4+ and CD8+ T cells 24-, 48- and 72-hours post allo-HCT. We then analyzed the expression of Gpr109a by qRT-PCR and compared it with unstimulated CD4+ and CD8+ T cells from naïve mice (Figure 2C). Expression of Gpr109a was highest at 24 hours and 48 hours post allo-activation.
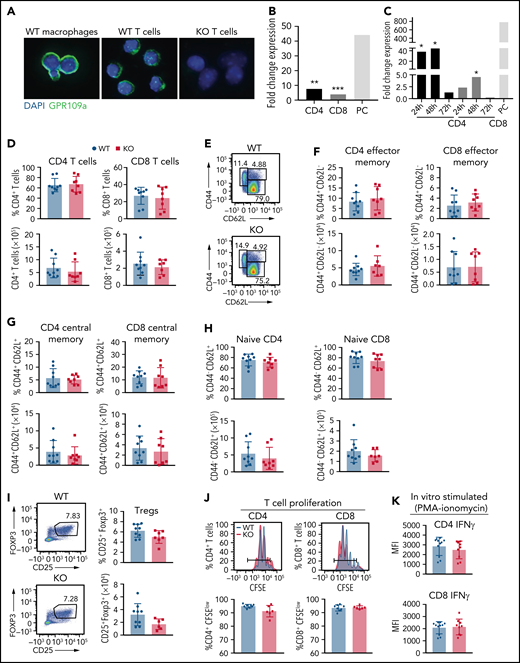
No difference in numbers and percentages of wild type and GPR109A−/− splenic T cells. (A) Immunofluorescence of magnetically sorted WT B6 macrophages, WT and KO T cells showing GPR109A (green) and DAPI (blue). Bar = 100 μm. (B) Magnetically sorted WT CD4+ and CD8+ T cells were stimulated with anti-CD3/CD28 beads for 72 hours and RNA was extracted to measure expression of GPR109A mRNA by qRT-PCR. Positive control (PC) was magnetically sorted F4-80+ cells. (C) FACS-sorted WT CD4+ and CD8+ T cells isolated from BALB/c irradiated mice transplanted with TCD BM and 10 × 106 WT T cells at 24-, 48-, and 72-hours post allo-HCT. RNA was extracted to measure expression of GPR109A mRNA by qRT-PCR and expression is shown as fold change compared with expression in naïve FACS-sorted CD4+ and CD8+ WT T cells, respectively. Expression levels were assessed as biological triplicates. Positive control (PC) was FACS sorted CD11b+ cells from a naïve BALB/c mouse spleen; expression of PC as fold change to naïve FACS-sorted CD4+ WT T cells. (D) Percentage and numbers of splenic CD4+ and CD8+ T cells from GPR109a+/+ (WT) and GPR109a−/− (KO) littermate mice at baseline. (E) Gating strategy for effector memory (CD44+CD62L−), central memory (CD44+CD62L+), and naïve (CD44−CD62L+) T cell populations. (F-H) Percent and numbers of effector memory (F), central memory (G), and naïve T cells (H), previously gated on live, CD45+, CD3+, and/or CD4+ and CD8+. (I) Representative flow cytometric analysis of percent and number of regulatory T cells (CD25+Foxp3+), previously gated on live, CD45+, CD3+, and CD4+. (J-K) Magnetically sorted WT and KO CD4+ and CD8+ T cells were labeled with CFSE and stimulated with anti-CD3/CD28 for 72 hours. Representative concatenated CFSE line graphs and quantification showing percentages of CFSEmedium/low WT and KO T cells. (K) CD4+ and CD8+ T cells were stimulated with PMA/ionomycin for 4 hours and mean fluorescence intensity (MFI) of intracellular IFNγ measured. Comparisons in (B) and (C) were performed by two-tailed unpaired Mann-Whitney Test versus unstimulated or naïve cells. *P < .05, **P < .01, ***P < .001. For (C) significance was calculated by comparing to naïve CD4+ or CD8+ T cells using a one-sample T-test. Differences in pairs in (D) to (K) are not significant, measured by two-tailed unpaired Mann-Whitney test, n = 6-10 mice per group. All values are means ± standard deviation. All results from three independent experiments.
No difference in numbers and percentages of wild type and GPR109A−/− splenic T cells. (A) Immunofluorescence of magnetically sorted WT B6 macrophages, WT and KO T cells showing GPR109A (green) and DAPI (blue). Bar = 100 μm. (B) Magnetically sorted WT CD4+ and CD8+ T cells were stimulated with anti-CD3/CD28 beads for 72 hours and RNA was extracted to measure expression of GPR109A mRNA by qRT-PCR. Positive control (PC) was magnetically sorted F4-80+ cells. (C) FACS-sorted WT CD4+ and CD8+ T cells isolated from BALB/c irradiated mice transplanted with TCD BM and 10 × 106 WT T cells at 24-, 48-, and 72-hours post allo-HCT. RNA was extracted to measure expression of GPR109A mRNA by qRT-PCR and expression is shown as fold change compared with expression in naïve FACS-sorted CD4+ and CD8+ WT T cells, respectively. Expression levels were assessed as biological triplicates. Positive control (PC) was FACS sorted CD11b+ cells from a naïve BALB/c mouse spleen; expression of PC as fold change to naïve FACS-sorted CD4+ WT T cells. (D) Percentage and numbers of splenic CD4+ and CD8+ T cells from GPR109a+/+ (WT) and GPR109a−/− (KO) littermate mice at baseline. (E) Gating strategy for effector memory (CD44+CD62L−), central memory (CD44+CD62L+), and naïve (CD44−CD62L+) T cell populations. (F-H) Percent and numbers of effector memory (F), central memory (G), and naïve T cells (H), previously gated on live, CD45+, CD3+, and/or CD4+ and CD8+. (I) Representative flow cytometric analysis of percent and number of regulatory T cells (CD25+Foxp3+), previously gated on live, CD45+, CD3+, and CD4+. (J-K) Magnetically sorted WT and KO CD4+ and CD8+ T cells were labeled with CFSE and stimulated with anti-CD3/CD28 for 72 hours. Representative concatenated CFSE line graphs and quantification showing percentages of CFSEmedium/low WT and KO T cells. (K) CD4+ and CD8+ T cells were stimulated with PMA/ionomycin for 4 hours and mean fluorescence intensity (MFI) of intracellular IFNγ measured. Comparisons in (B) and (C) were performed by two-tailed unpaired Mann-Whitney Test versus unstimulated or naïve cells. *P < .05, **P < .01, ***P < .001. For (C) significance was calculated by comparing to naïve CD4+ or CD8+ T cells using a one-sample T-test. Differences in pairs in (D) to (K) are not significant, measured by two-tailed unpaired Mann-Whitney test, n = 6-10 mice per group. All values are means ± standard deviation. All results from three independent experiments.
To assess whether GPR109A deficiency had an impact on T cell development, we characterized splenic T cells from WT and KO littermates at baseline by flow cytometry (Figure 2D-I). We observed no differences in frequency or absolute counts of CD4+ and CD8+ T cells (Figure 2D), effector memory (Figure 2E,F), central memory (Figure 2G), and naïve (Figure 2H) T cells. There was no difference in frequency or absolute counts of regulatory T cells (Tregs) at baseline (Figure 2I). WT and KO T cells proliferated at similar rates (Figure 2J), and KO T cells exhibited a comparable capacity to produce IFNγ after PMA-ionomycin stimulation in vitro (Figure 2K). We also differentiated naïve CD4 T cells in vitro into T cell subsets, Th1, Th2, Th17, and Tregs, and observed similar frequencies for WT and KO T cells in all conditions tested (supplemental Figure 3A-D). Additionally, in an in vitro Treg suppression assay, WT and KO Tregs suppressed responder T cells to the same extent (supplemental Figure 3E).
To examine if there were transcriptional differences between WT and KO CD4+ T cells, we FACS-sorted unstimulated or in vitro stimulated (for 72 hours with anti-CD3/CD28) T cells and submitted replicates for RNA sequencing analysis. There were few transcriptional differences between unstimulated and stimulated WT and KO CD4+ T cells observed by principal component analysis (PCA) (supplemental Figure 4A) and differential expression analysis (supplemental Figure 4B-C). Taken together, the absence of GPR109A on T cells does not alter the transcriptional profile, baseline frequency, number of splenic T cells, or their capacity to differentiate, proliferate, polarize, or produce IFNγ.
GPR109A-deficient T cells induce less GVHD organ pathology and inflammation
To further understand the differences in GVHD morbidity and mortality in allo-HCT recipients of GPR109A-deficient donor T cells, we analyzed GVHD target-organ pathology in the MHC-disparate model (B6 to BALB/c). Histological examination of GVHD target organs (skin, liver, small intestine, and colon) revealed significantly lower total GVHD scores on a validated scale27 in recipients of KO T cells at day 7 and day 14 post allo-HCT (Figure 3B and supplemental Figure 5A). Allo-HCT recipients of KO T cells had significantly lower GVHD histopathology scores in the liver and skin on day 7 (supplemental Figure 5A) and in all target organs at day 14 (representative histopathological micrographs shown in Figure 3A-B).
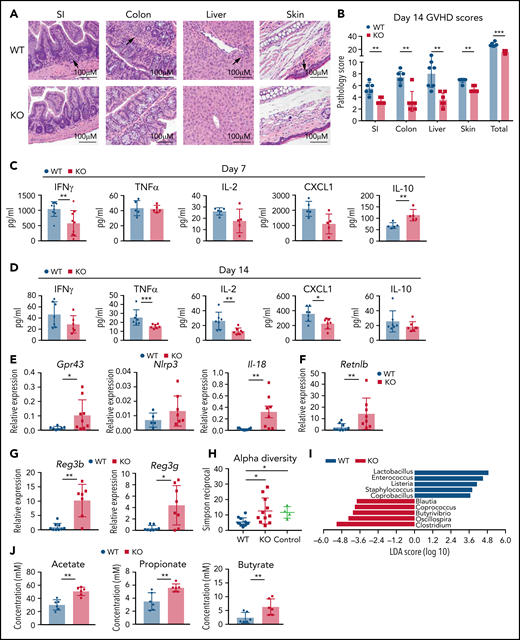
GPR109A−/− T cell recipients have reduced pathology and inflammation. Lethally irradiated BALB/c recipients received B6 WT TCD BM and 0.5 × 106 WT or KO T cells. (A) Representative H&E images of GVHD target organs small intestine (SI), large intestine, liver and skin on day 14 after transplant. Arrows indicate high density of infiltrating lymphocytes. (B) Organs were scored for histopathalogic damage at day 14. (C-D) Serum cytokines were measured at 7 days post allo-HCT (C) and 14 days post allo-HCT (D). (E-G) mRNA was extracted from colonic tissue at day 7 post allo-HCT to measure expression of specific intestinal genes. (H-I) Bacterial DNA was extracted from day 7 stool and 16S gene amplicon sequencing to measure alpha diversity (H) and linear discriminate analysis of effect size (LEfSe) analysis comparing bacterial differences between WT and KO T cell recipients (I). Controls are stool collected from WT BALB/c mice. (J) SCFAs from day 7 cecal contents was measured by GC-MS. All comparisons in (B) to (J) were performed by two-tailed unpaired Mann-Whitney Test. Values are means ± standard deviation. *P < .05, **P < .01, ***P < .0001, n = 5-10 mice per group. All results from two independent experiments.
GPR109A−/− T cell recipients have reduced pathology and inflammation. Lethally irradiated BALB/c recipients received B6 WT TCD BM and 0.5 × 106 WT or KO T cells. (A) Representative H&E images of GVHD target organs small intestine (SI), large intestine, liver and skin on day 14 after transplant. Arrows indicate high density of infiltrating lymphocytes. (B) Organs were scored for histopathalogic damage at day 14. (C-D) Serum cytokines were measured at 7 days post allo-HCT (C) and 14 days post allo-HCT (D). (E-G) mRNA was extracted from colonic tissue at day 7 post allo-HCT to measure expression of specific intestinal genes. (H-I) Bacterial DNA was extracted from day 7 stool and 16S gene amplicon sequencing to measure alpha diversity (H) and linear discriminate analysis of effect size (LEfSe) analysis comparing bacterial differences between WT and KO T cell recipients (I). Controls are stool collected from WT BALB/c mice. (J) SCFAs from day 7 cecal contents was measured by GC-MS. All comparisons in (B) to (J) were performed by two-tailed unpaired Mann-Whitney Test. Values are means ± standard deviation. *P < .05, **P < .01, ***P < .0001, n = 5-10 mice per group. All results from two independent experiments.
To analyze differences in the development of GVHD between recipients of WT T cells vs KO T cells, we measured serum concentrations of inflammatory cytokines IFNγ, TNFα, IL-2, IL-6, IL-10, CXCL1, macrophage inflammatory protein 2 (MIP-2), IL-17, and IL-12p70 (Figure 3C-D; supplemental Figure 5B-C), which have been implicated in the pathophysiology of acute GVHD.33,34 We observed significantly increased serum concentrations of IL-10, decreased IFNγ at day 7 (Figure 3C), and TNFα, IL-2, and CXCL1 at day 14 (Figure 3D) in KO T-cell recipients.
As the gut is a major target organ for GVHD, we further analyzed gut mucosal changes and overall gut health in more detail by measuring transcript abundance of several genes by qRT-PCR in colonic tissue on day 7 post allo-HCT (Figure 3E-G; supplemental Figure 5D-K). In recipients of KO T cells, we measured significantly higher transcript abundance of Gpr43 and Il-18 in colonic tissues (Figure 3E). Recipients of KO T cells also had increased relative expression of the goblet-cell-derived gene resistin-like molecule β (Retnlb) (Figure 3F), which is important for the regulation of intestinal colitis.35,36 The Paneth cell protein regenerating islet-derived 3γ (Reg3γ) can counteract GI-GVHD in mice,37 and expression of the genes encoding both antimicrobial peptides (AMPs) Reg3b and Reg3g were increased in KO T-cell recipient colonic tissue (Figure 3G). The higher expression of AMP transcripts led us to hypothesize that the microbiota perturbations induced by GVHD might be attenuated in recipients of KO T cells.37,38 Microbiota profiling by 16S rRNA gene sequencing revealed less microbial community disruption post allo-HCT in recipients of KO T cells, indicated by increased overall α diversity measured by the Simpson reciprocal index (Figure 3H). We further assessed the taxa that best explained the differences between the 2 groups using linear discriminate analysis of effect size (LEfSe) (Figure 3I). Gram-positive bacteria, including Lactobacillus and Enterococcus, which are targets for both Reg3β and Reg3γ AMPs, were increased in WT T-cell recipients. Enterococcus has been associated with increased GVHD in both patients and mouse models,27,39 and we have previously demonstrated an increase in Lactobacillus in mice with GVHD.20 Conversely, KO T-cell recipients exhibited an increased abundance of the taxa Clostridium and Blautia, which are associated with reduced GVHD-related mortality.40 Furthermore, recipients of KO T cells had higher cecal concentrations of the SCFAs acetate, propionate, and butyrate (Figure 3J). These results demonstrate that compared with recipients of WT T cells, KO T-cell recipients display significantly less target-organ GVHD, lower circulating concentrations of inflammatory cytokines, less intestinal damage, and less GVHD-associated perturbations of host-microbiome homeostasis.20,37,41
Allo-HCT recipients with GVHD have fewer GPR109A-deficient T cells
To study the mechanism by which GPR109A contributes to alloreactivity of T cells, we characterized immune cell populations in the spleen and lamina propria (LP) of small intestine (SI) and colon tissues in allo-HCT recipients. We observed significantly lower numbers of donor CD4+ T cells in all 3 tissues at day 7 and day 14 in recipients of KO T cells (Figure 4A) compared with recipients of WT T cells. Specifically, we found significant decreases in CD4+ effector memory T cells in the indicated tissues at both time points (Figure 4B). We also observed decreased numbers of donor CD8+ T cells in SI and colon LP at day 7, and decreased numbers of CD8+ T cells in spleen and SI LP at day 14 (Figure 4C), as well as decreased CD8+ effector memory T cells in all 3 tissues, particularly at day 14 (Figure 4D). We found an increase in the percentage of Tregs at days 7 and 14, along with an increase in the ratio of Tregs to conventional CD4+ T cells in recipients of KO T cells (Figure 4E-F). Differentiation to Th1 or Th17 was equivalent between KO and WT T cells with the exception of small but statistically significant decreases in CD4+T-bet+ cells in the spleen and CD4+Rorgt+ T cells in the spleen and SI (Figure 4G-H).
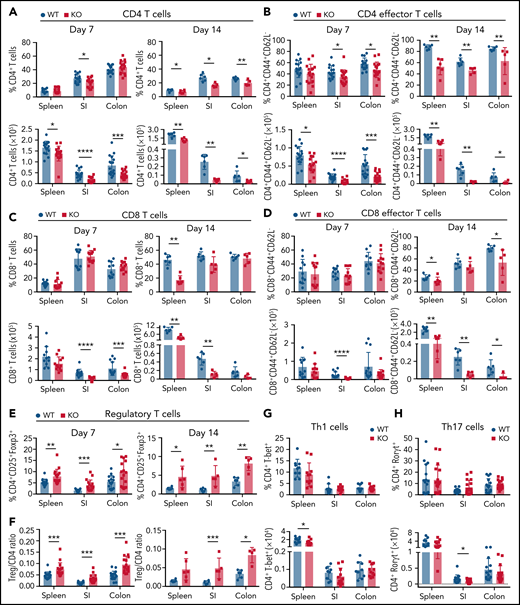
Recipients of GPR109A−/− T cells have fewer donor T cells post allo-HCT compared to WT. Lethally irradiated BALB/c recipients received B6 WT TCD BM and 0.5 × 106 WT or KO T cells. (A-B) Live, CD45+, CD3+, H-2kb+ donor CD4+ and CD8+ T cell percentages (top) and numbers (bottom) are shown from spleen, small intestine (SI) lamina propria and colon lamina propria on day 7 and day 14 post allo-HCT. (C-D) Live, CD45+, CD3+, H-2kb+ donor effector memory CD4+ and CD8+ T cell percentages and numbers were analyzed from indicated tissues. (E) Live, CD45+, CD3+, H-2kb+, CD4+ donor regulatory T cell percentages analyzed for the 3 tissues on day 7 and 14 and (F) ratio of Tregs to CD4+ T cells analyzed. (G) Percent and number of T-bet+ Th1 cells on day 7 post-BMT. (H) Percent and number of Rorgt+ Th17 cells on day 7 post allo-HCT. All comparisons in (A) to (H) were performed by two-tailed unpaired Mann-Whitney Test. Values are means ± standard deviation. *P < .05, **P < .01, ***P < .001, ****P < .0001, n = 5-15 mice per group. All results from two to three independent experiments.
Recipients of GPR109A−/− T cells have fewer donor T cells post allo-HCT compared to WT. Lethally irradiated BALB/c recipients received B6 WT TCD BM and 0.5 × 106 WT or KO T cells. (A-B) Live, CD45+, CD3+, H-2kb+ donor CD4+ and CD8+ T cell percentages (top) and numbers (bottom) are shown from spleen, small intestine (SI) lamina propria and colon lamina propria on day 7 and day 14 post allo-HCT. (C-D) Live, CD45+, CD3+, H-2kb+ donor effector memory CD4+ and CD8+ T cell percentages and numbers were analyzed from indicated tissues. (E) Live, CD45+, CD3+, H-2kb+, CD4+ donor regulatory T cell percentages analyzed for the 3 tissues on day 7 and 14 and (F) ratio of Tregs to CD4+ T cells analyzed. (G) Percent and number of T-bet+ Th1 cells on day 7 post-BMT. (H) Percent and number of Rorgt+ Th17 cells on day 7 post allo-HCT. All comparisons in (A) to (H) were performed by two-tailed unpaired Mann-Whitney Test. Values are means ± standard deviation. *P < .05, **P < .01, ***P < .001, ****P < .0001, n = 5-15 mice per group. All results from two to three independent experiments.
We examined the role of CD8+ T cells by investigating cytotoxic markers in recipients of WT or KO T cells (supplemental Figure 6). At day 7 post allo-HCT, there were minor differences in the number of WT vs KO CD8+ Granzyme B+ T cells (supplemental Figure 6A). On day 14, we observed an increased frequency and number of KO CD8+ Granzyme B+ T cells as well as increased numbers of CD8+CD107+ KO T cells (supplemental Figure 6B). In addition, we observed in an in vitro CTL assay that both WT and KO T cells induced similar levels of cell lysis (supplemental Figure 6C).
Alloreactive Gpr109a−/− T cells undergo increased apoptosis
To understand why there were fewer T cells in recipients of KO grafts at day 7 and day 14, we studied the early kinetics of T-cell activation and proliferation, focusing on the spleen and mesenteric lymph nodes (mLNs). Lower numbers of donor T cells in recipients of KO T cells were already apparent at day 3 post allo-HCT in the spleen (Figure 5A) and mLNs (supplemental Figure 7A), as well as an increased percentage of donor Tregs in spleens of KO T-cell recipients (Figure 5B).
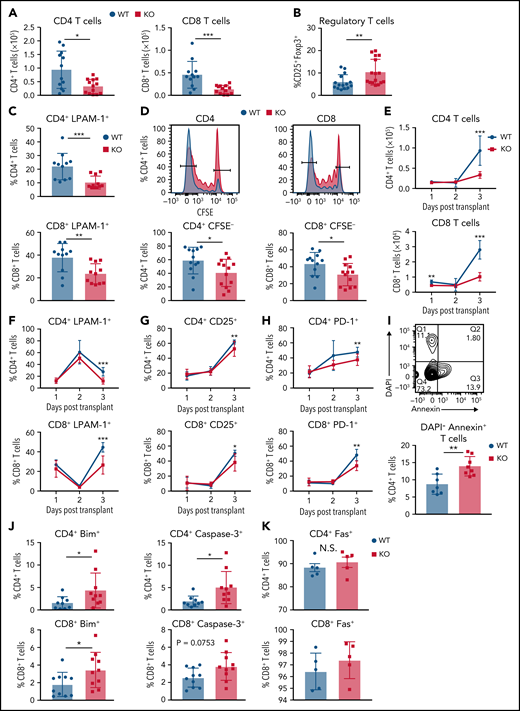
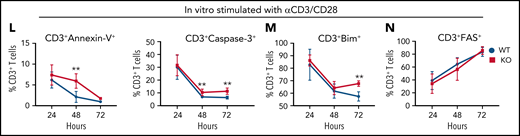
GPR109A−/− T cells undergo increased apoptosis. Lethally irradiated BALB/c recipients received B6 WT TCD BM and 5 × 106 WT or KO T cells. (A) Donor CD4+ and CD8+ T cell numbers were analyzed from spleen on day 3 post allo-HCT. (B) Donor regulatory T cells from spleen on day 3 post allo-HCT. (C) Expression of LPAM-1 on splenic donor CD4+ and CD8+ T cells on day 3 post allo-HCT. (D) T cells were labeled with CFSE prior to transplant and proliferation determined by measuring CFSElow cells. (E-H) Early timecourse at day 1, 2, and 3 post allo-HCT measuring numbers of CD4+ and CD8+ T cells (E), LPAM-1+ CD4+ and CD8+ T cells (F) CD4+ and CD8+ T cells expressing activation markers, CD25 (G) and PD-1 (H). (I) Frequency of Annexin+DAPI- apoptotic donor CD4+ T cells were analyzed on day 3 post alloHCT. (J-K) Frequency of donor CD4+ (top) and CD8+ (bottom) T cells on day 3 post allo-HCT, expressing other apoptotic markers Bim, cleaved Caspase-3 (J) and Fas(K). (L-N) MACS sorted splenic T cells were stimulated in vitro with anti-CD3/CD28 for 24, 48, or 72 hours and frequency of apoptotic cells measured by flow cytometry gating on live CD3+ and Annexin+DAPI- (L), cleaved caspase-3+, Bim+ (M) or Fas+ (N). All comparisons in (A) to (M) were performed by two-tailed unpaired Mann-Whitney Test. Values are means ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001, n = 15 mice per group. All results from two to three independent experiments.
GPR109A−/− T cells undergo increased apoptosis. Lethally irradiated BALB/c recipients received B6 WT TCD BM and 5 × 106 WT or KO T cells. (A) Donor CD4+ and CD8+ T cell numbers were analyzed from spleen on day 3 post allo-HCT. (B) Donor regulatory T cells from spleen on day 3 post allo-HCT. (C) Expression of LPAM-1 on splenic donor CD4+ and CD8+ T cells on day 3 post allo-HCT. (D) T cells were labeled with CFSE prior to transplant and proliferation determined by measuring CFSElow cells. (E-H) Early timecourse at day 1, 2, and 3 post allo-HCT measuring numbers of CD4+ and CD8+ T cells (E), LPAM-1+ CD4+ and CD8+ T cells (F) CD4+ and CD8+ T cells expressing activation markers, CD25 (G) and PD-1 (H). (I) Frequency of Annexin+DAPI- apoptotic donor CD4+ T cells were analyzed on day 3 post alloHCT. (J-K) Frequency of donor CD4+ (top) and CD8+ (bottom) T cells on day 3 post allo-HCT, expressing other apoptotic markers Bim, cleaved Caspase-3 (J) and Fas(K). (L-N) MACS sorted splenic T cells were stimulated in vitro with anti-CD3/CD28 for 24, 48, or 72 hours and frequency of apoptotic cells measured by flow cytometry gating on live CD3+ and Annexin+DAPI- (L), cleaved caspase-3+, Bim+ (M) or Fas+ (N). All comparisons in (A) to (M) were performed by two-tailed unpaired Mann-Whitney Test. Values are means ± standard deviation. *P < 0.05, **P < 0.01, ***P < 0.001, n = 15 mice per group. All results from two to three independent experiments.
We speculated that the reduced numbers of KO T cells at this early time point could be due to less trafficking from the systemic circulation to the gut via the mLNs. LPAM-1 is a critical homing integrin necessary for the development of intestinal GVHD.42 We found a significant decrease in the frequency of splenic (Figure 5C) and mLN (supplemental Figure 7B) LPAM-1+ T cells in KO T-cell recipients. We did not detect any difference in proliferation of WT vs KO T cells after in vitro anti-CD3/CD28 stimulation (Figure 2I). However, we found that T cells in KO T-cell recipients proliferated less in vivo compared with WT (Figure 5D).
To determine if there was an initial early expansion of KO T cells followed by a contraction, representing recipient DC-mediated activation-induced cell death (AICD), we measured numbers of CD4+ and CD8+ T cells at 1, 2, and 3 days following allo-HCT (Figure 5E-H; supplemental Figure 7C-D). CD4+ KO T cells were present in similar numbers to WT on day 1 and day 2, with decreased numbers by day 3 post allo-HCT in both spleen and mLNs. There were reduced frequency of LPAM-1+ T cells in KO T-cell recipients on day 3 post allo-HCT in the spleen (Figure 5F) and mLNs (supplemental Figure 7D). We also measured the frequency of activated T cells using activation markers CD25 and PD-1 in the spleen (Figure 5G-H) and mLNs (supplemental Figure 7E-F). We observed significant differences in frequencies of both CD25+ and PD-1+ KO T cells in the spleens of allo-HCT recipients on day 3, indicating reduced activation of KO T cells. To further analyze activation of WT and KO T cells, we noted reduced expression of pp-Erk1/2 in KO T-cell recipient T cells at day 3 post allo-HCT, as measured by mean fluorescence intensity (MFI) (supplemental Figure 7G) as well as reduced expression of phosphorylated Akt (pAkt) and pp-ERK1/2 at day 7 post allo-HCT (supplemental Figure 7H). In summary, KO T cells become less activated compared with WT post allogeneic stimulation.
We hypothesized that the reduction in cell numbers and decreased activation of KO T cells could be due to exhaustion or cell death. However, KO T cells did not show signs of T-cell exhaustion in any of the tissues examined after allo-HCT (supplemental Figure 8A-B). We further analyzed cell death and observed an increased percentage of Annexin V+ splenic CD4+ T cells in KO T-cell recipients (Figure 5I). Two other proapoptotic markers, Bim and cleaved Caspase-3, were also increased in CD4+ and CD8+ KO T cells (Figure 5J) and CD4+ KO T cells in mLNs (supplemental Figure 7I) after transplant. However, the expression of FAS, a protein typically triggered in AICD,43 was not different between WT and KO CD4+ or CD8+ T cells (Figure 5K), which may suggest mitochondria-induced apoptosis.44 In an in vitro time-course experiment to measure the percentage of T cells expressing apoptotic markers 24, 48, and 72 hours after stimulation with anti-CD3/CD28, we found that by 48 hours, there was a significantly increased frequency of KO T cells expressing the apoptotic markers Annexin-V and cleaved Caspase-3 compared with WT (Figure 5L). By 72 hours, there was a higher frequency of Bim+ KO T cells compared with WT (Figure 5M), and there was no difference in expression of FAS at any time point post in vitro stimulation (Figure 5N).
Alloreactive KO T cells become metabolically dysregulated
To understand whether butyrate differentially affects WT vs KO T cell after immune stimulation, we stimulated CD4+ and CD8+ T cells in vitro with anti-CD3/CD28 and increasing doses of butyrate (10 µM, 100 µM, and 1000 µM) that are equivalent to in vivo plasma or tissue levels16,45 (supplemental Figure 9A-B). Butyrate significantly inhibited proliferation at higher doses, most likely due to toxic effects of the SCFA as reported32,46 (supplemental Figure 9C-D), and this effect was independent of the genotype. In an in vitro MLR assay with WT or KO T cells plus/minus 100 µM butyrate (supplemental Figure 9E-J), we saw a significant decrease in numbers of KO CD4+ T cells compared with WT T cells, but this was again independent of butyrate (supplemental Figure 9E). There was minimal difference in divided CD4+ KO T cells (supplemental Figure 9F), and the percentage of apoptotic Bim+ KO CD4+ T cells was increased, but butyrate did not affect the responses (supplemental Figure 9G). Similar phenotypes, again independent of butyrate, were observed for WT and KO CD8+ T cells (supplemental Figure 9H-J).
Production of reactive oxygen species (ROS) is critical for the metabolic programming of T cells, but high concentrations of ROS in T cells can trigger programmed cell death.47 We observed an increased level of intracellular ROS in KO T cells on day 3 post allo-HCT (Figure 6A). To further investigate our hypothesis that apoptosis of KO T cells occurs due to activation of mitochondrial apoptosis pathways and may be due to dysregulation of intracellular metabolic pathways, we measured mitochondrial respiratory function of unstimulated and stimulated T cells from WT and KO littermates. We observed no difference in basal oxygen consumption rate (OCR) at baseline between unstimulated T cells (supplemental Figure 9K-L). However, after 72 hours of in vitro stimulation with anti-CD3/CD28, KO T cells had significantly decreased basal OCR and spare respiratory capacity compared with WT T cells (Figure 6B). We also examined mitochondrial membrane potential of in vitro stimulated, purified WT and KO T cells using the cationic JC-1 dye.48 Activated KO T cells had a decreased red/green fluorescence ratio, reflecting increased mitochondrial depolarization and reduced metabolic activity (Figure 6C). We hypothesized that the metabolic dysfunction of KO T cells could potentially be reversed by adding the antioxidant N-acetyl cysteine (NAC) because treatment of T cells with NAC can reduce apoptosis, induce proliferation, and enhance antitumor immunity.49,50 We returned to in vivo GVHD experiments in which the drinking water of BALB/c recipients of WT or KO donor T cells were supplemented or not with NAC. While NAC treatment had no effect on GVHD survival in recipients of WT T cells, strikingly, this treatment rescued the alloreactivity of KO T cells (Figure 6D). This indicates that improving the metabolic health of KO T cells restored their alloreactive potential and GVHD activity.
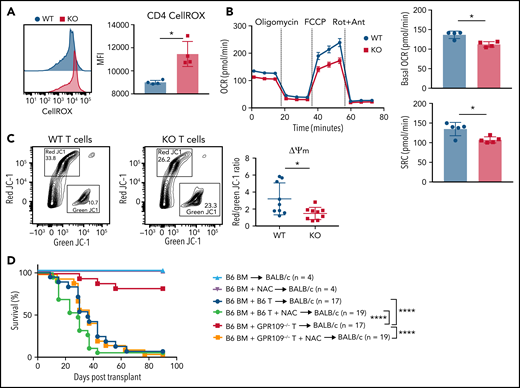
GPR109A−/− T cells become metabolically dysfunctional after activation. (A) MFI of CellROX green in WT and KO T cells on day 3 post allo-HCT. (B) OCR of in vitro activated WT (black) or KO (red) T cells, as measured under basal conditions and in response to oligomycin, FCCP, and Rot + Ant and spare respiratory capacity of WT and KO T cells. (C) Representative flow cytometric analysis of green vs. red JC-1 aggregates of in vitro activated WT and KO purified T cells without (blue) and with (green) N-acetylcysteine (NAC) in the drinking water, or WT BM with KO T cells without (red) and with (orange) NAC in the drinking water. Controls were transplanted with T cell-depleted BM only without (light blue) and with NAC (purple) in the drinking water. Comparisons in (H) were performed by Mantel-Cox log rank test for survival. All comparisons in (A) to (C) were performed by two-tailed unpaired Mann-Whitney Test. Values are means ± standard deviation. *P < .05, ****P < .0001, n = 4-9 mice per group, or as indicated. All results from two independent experiments.
GPR109A−/− T cells become metabolically dysfunctional after activation. (A) MFI of CellROX green in WT and KO T cells on day 3 post allo-HCT. (B) OCR of in vitro activated WT (black) or KO (red) T cells, as measured under basal conditions and in response to oligomycin, FCCP, and Rot + Ant and spare respiratory capacity of WT and KO T cells. (C) Representative flow cytometric analysis of green vs. red JC-1 aggregates of in vitro activated WT and KO purified T cells without (blue) and with (green) N-acetylcysteine (NAC) in the drinking water, or WT BM with KO T cells without (red) and with (orange) NAC in the drinking water. Controls were transplanted with T cell-depleted BM only without (light blue) and with NAC (purple) in the drinking water. Comparisons in (H) were performed by Mantel-Cox log rank test for survival. All comparisons in (A) to (C) were performed by two-tailed unpaired Mann-Whitney Test. Values are means ± standard deviation. *P < .05, ****P < .0001, n = 4-9 mice per group, or as indicated. All results from two independent experiments.
Discussion
In the current study, we describe a novel role for the butyrate receptor GPR109A in alloreactive T cells in GVHD. At steady state, naïve, memory, and Tregs compartments are present in equal numbers and frequency in Gpr109a KO vs WT mice; they showed similar proliferative capacity and ability to produce IFNγ. This finding aligns with a previous report that WT and KO splenic CD4+ T cells produced equivalent amounts of IL-17 and IL-10 at steady state.31 In the setting of mouse GVHD, the absence of GPR109A in recipient tissues had no significant effect on survival. This could be due to compensatory function by another SCFA receptor, such as GPR43 or the butyrate transporter SLC5a8. However, GPR109A was required by T cells in the transplant graft to induce GVHD morbidity and mortality, as well as target organ pathology. This finding was accompanied by an attenuation of GVHD-induced changes to the intestinal microbiome. In KO T-cell recipient spleens, CD8+ T cells had increased expression of granzyme B and the degranulation marker CD107, in spite of less GVHD. However, there was a significantly decreased overall number of CD8+ T cells present in the KO T-cell recipient spleens, and this model of GVHD is primarily driven by CD4+ T cells.51 Therefore, we hypothesize that despite the increased activation of CD8+ KO T cells, this was likely not enough to cause a difference in GVHD. Overall, KO donor T cells were less activated, exhibited decreased expression of a gut-homing molecule, LPAM-1, and displayed increased apoptosis. Nevertheless, the apoptosis observed appeared to be mitochondrially-induced as evidenced by the lack of expansion and contraction, as well as increased expression of proapoptotic markers Bim and Caspase-3, with no difference in FAS upregulation. These findings are in line with previous studies showing that GPR109A plays a role in the metabolic regulation of cells in which it is expressed; however, this is the first time it has been described in T cells.52-54
GPCRs can regulate several cellular processes in T cells, including homeostatic proliferation, cell size, and metabolism, either through direct downstream signaling or by guiding T-cell migration toward specific signals.55 It is possible that GPR109A metabolically stabilizes T cells, allowing them to receive necessary signals such as LPAM-1 to migrate to the gut. Our data are consistent with a model in which T cells become metabolically unstable in the absence of GPR109A, leading to increased apoptosis and, therefore, less allo-activation. On the other hand, KO T cells were still capable of responding to the same extent as WT T cells in a viral infection and to allogeneic lymphoma cells in the models tested here.
The role of GPR109A signaling in T cells has not previously been well studied. At high concentrations (>1 mM), butyrate induces cell death of T cells independent of GPR109A (data not shown), which has been described for T cells and other immune cell types.32,46 Depending on the concentration, butyrate can induce FAS-mediated apoptosis or Th1-associated factors IFNγ and T-bet, independent of GPRs.56 Butyrate was also shown to promote memory potential of activated CD8+ T cells through effects on cellular metabolism,57 highlighting the many roles of butyrate in T cell function and regulation. In the setting of GVHD, butyrate is a survival factor for enterocytes16 and protects from GVHD on nonhematopoietic target tissues of the host in a dose-dependent manner via GPR43.23
For the first time, we provide evidence that GPR109A in allo-activated T cells plays an important role in the pathogenesis of GVHD. The observed decrease in GVHD is likely a secondary effect of fewer Gpr109a−/− T cells, leading to decreased epithelial cell damage in the gut, increased production of antimicrobial peptides, and increased abundance of SCFA-producing bacteria. Notably, Gpr109a−/− T cells induce less GVHD in the host but still retain antitumor as well as antiviral activity in a nonallogeneic setting. Our findings on GPR109A on T cells call for further evaluation in pharmacological and clinical translational studies in the allo-HCT setting. Prior to transplant, T cells could be modified to no longer express GPR109A either through the use of short-hairpin RNA (sh-RNA), or a neutralizing antibody targeting GPR109A could be used. Furthermore, transplant donors and recipients could be tested for gene variations at the GPR109A coding regions prior to transplant and followed up for GVHD outcomes.
Acknowledgments
J.U.P. reports funding from NHLBI NIH Award K08HL143189, the MSKCC Cancer Center Core Grant NCI P30 CA008748. This research was supported by the Parker Institute for Cancer Immunotherapy at Memorial Sloan Kettering Cancer Center, J.U.P is a member of the Parker Institute for Cancer Immunotherapy. M.R.M.v.d.B. reports funding by National Cancer Institute award numbers, R01-CA228358, R01-CA228308, R01-HL147584, P30 CA008748 MSK Cancer Center Support Grant/Core Grant, and Project 2 of P01-CA023766, National Heart, Lung, and Blood Institute (NHLBI) award numbers R01-HL125571 (A. Hanash) and R01-HL123340 (K. Cadwell), National Institute of Aging award number Project 2 of P01-AG052359 (J. Nikolich-Zugich/M.v.d.B.), National Institute of Allergy and Infectious Diseases award number U01 AI124275. Additional funding was received from The Lymphoma Foundation, The Susan and Peter Solomon Divisional Genomics Program, Starr Cancer Consortium, Tri-Institutional Stem Cell Initiative, Cycle for Survival, and the Parker Institute for Cancer Immunotherapy at Memorial Sloan Kettering Cancer Center.
The authors thank Klaus Pfeffer (University of Dusseldorf) for providing the Gpr109a−/− mice and Ikuo Kimura (Kyoto University) for providing the Gpr43−/− mice.
Authorship
Contribution: M.D.D. designed and performed experiments and wrote the manuscript; M.B.d.S., A.L., K.B.N., S.R.L., G.K.A., A.E.S., Y.S., C.N, S.M., E.D., N.L., C.D.G., S.M.P., M.S., I.G., J.R.C., and C.K.S.-T. performed experiments, edited the manuscript, and provided expertise and feedback; A.I.K. and E.Z. analyzed data, edited the manuscript, and provided expertise and feedback; K.A.M., S.A.V., J.C.S., J.U.P., and R.R.J. edited the manuscript and provided expertise and feedback; and M.R.M.v.d.B. edited the manuscript, provided expertise and feedback, and secured funding.
Conflict-of-interest disclosure: J.U.P. reports research funding, intellectual property fees, and travel reimbursement from Seres Therapeutics and consulting fees from DaVolterra, CSL Behring, and MaaT Pharma. J.U.P. has filed intellectual property applications related to the microbiome (reference numbers #62/843 849, #62/977 908, and #15/756 845). M.R.M.v.d.B has received research support and stock options from Seres Therapeutics and stock options from Notch Therapeutics and Pluto Therapeutics; has received royalties from Wolters Kluwer; has consulted, received honorarium from or participated in advisory boards for Seres Therapeutics, WindMIL Therapeutics, Rheos Medicines, Merck & Co, Inc., Magenta Therapeutics, Frazier Healthcare Partners, Nektar Therapeutics, Notch Therapeutics, Forty Seven Inc., Priothera, Ceramedix, Lygenesis, Pluto Therapeutics, GlaskoSmithKline, Da Volterra, Vor BioPharma, Novartis (spouse), Synthekine (spouse), and Beigene (spouse); has IP licensing with Seres Therapeutics and Juno Therapeutics; and holds a fiduciary role on the Foundation Board of DKMS (a nonprofit organization). The remaining authors declare no competing financial interests.
Correspondence: Marcel R. M. van den Brink, Department of Hematologic Malignancies, Memorial Sloan Kettering Cancer Center, 417 East 68th Street, 14th Floor, New York, NY 10065; e-mail: Vandenbm@mskcc.org.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal